Cytoskeletal Blueprint of Invasion: Decoding Architectural Cues in Cancer Cells for Diagnostic and Therapeutic Advancements
This article provides a comprehensive analysis of the distinct cytoskeletal architectures that differentiate invasive from non-invasive cancer cells, tailored for researchers and drug development professionals.
Cytoskeletal Blueprint of Invasion: Decoding Architectural Cues in Cancer Cells for Diagnostic and Therapeutic Advancements
Abstract
This article provides a comprehensive analysis of the distinct cytoskeletal architectures that differentiate invasive from non-invasive cancer cells, tailored for researchers and drug development professionals. It explores the foundational role of cytoskeletal dynamics in cell invasion, evaluates advanced computational and live-cell imaging methodologies for quantitative analysis, addresses key technical challenges in assay systems, and validates cytoskeletal targets for therapeutic intervention. By synthesizing recent findings on microtubule organization, actin dynamics, and cytoskeletal crosslinkers like plectin, this review establishes the cytoskeleton as a critical biomarker and a promising target for anti-metastatic strategies, offering a roadmap for future biomedical and clinical research.
Architectural Hallmarks: How Cytoskeletal Reorganization Drives the Invasive Phenotype
The cytoskeleton is a dynamic, three-dimensional network essential for maintaining cellular architecture and enabling critical processes such as cell division, migration, and invasion [1]. During cancer progression, cells undergo profound morphological transformations, and the reorganization of the cytoskeleton, particularly microtubules, is known to play a key role [1]. Microtubules, composed of α-/β-tubulin heterodimers, are not merely structural elements; their precise organization—including their quantity, orientation, compactness, and radiality—creates identifiable signatures that can distinguish invasive from non-invasive cellular phenotypes [1]. The ability to detect and quantify these subtle architectural alterations provides a powerful proxy for identifying aggressive cancer cells at an early stage, addressing a critical limitation in current diagnostic approaches [1]. This guide objectively compares the microtubule signatures associated with invasive potential, summarizing key experimental data and providing the methodological toolkit for researchers and drug development professionals to apply these findings in their work.
Quantitative Comparison of Microtubule Signatures
The architectural features of microtubules undergo distinct reorganization in cells with invasive potential. The quantitative differences outlined in the table below provide a signature profile for identifying such cells.
Table 1: Quantitative Microtubule Signatures in Non-Invasive vs. Invasive Cells
| Microtubule Feature | Non-Invasive Cell Signature | Invasive Cell Signature | Measurement Significance |
|---|---|---|---|
| Fiber Orientation | Higher Orientational Order Parameter (OOP) [1] | Lower OOP value, indicating disperse fiber orientations [1] | Measures global alignment of fibers; lower OOP signifies disorganization. |
| Fiber Length | Longer, more stable microtubules [1] | Shorter microtubules [1] | Indicates microtubule polymerization dynamics and stability. |
| Fiber Quantity (Nl) | Variable, dependent on cell state [1] | Variable, dependent on cell state [1] | The absolute number of polymerized microtubule fibers in a cell. |
| Fiber Compactness | Lower fiber density (e.g., 0.421 μm⁻²) [1] | Higher fiber density (e.g., 1.539-2.039 μm⁻²) [1] | Number of fibers per unit area (Nl/Ac); indicates spatial distribution. |
| Radiality (Radial Score) | Lower radial score (e.g., 0.266) [1] | Prominent radial pattern from nucleus (e.g., 0.564) [1] | Measures the extent to which fibers nucleate from the nucleus centroid. |
These signatures were validated in a model of cells expressing wild-type E-cadherin (non-invasive) versus a mutant E-cadherin that leads to loss of cell-cell adhesion and an invasive phenotype. The analysis confirmed that mutant, invasive cells exhibit significantly lower OOP values, corresponding to a more disorganized microtubule network [1].
Experimental Protocols for Microtubule Signature Analysis
Computational Pipeline for Cytoskeletal Architecture
A novel computational pipeline has been developed to dissect the cytoskeletal architecture of cancer cells with invasive potential from immunofluorescence images [1]. The following workflow details the key steps:
Table 2: Key Experimental Reagents and Solutions
| Research Reagent | Function / Application |
|---|---|
| α-Tubulin Antibody | Immunofluorescence staining to visualize the microtubule network. |
| Cell Culture with Laminin | Provides a supportive extracellular matrix environment for cell growth. |
| E-cadherin Mutant Model | A well-established cellular model (e.g., p.L13_L15del mutant) that leads to loss of cell-cell adhesion and an invasive phenotype for validation. |
| Deconvolution Software | Removes noise and blur from Z-stack images, improving contrast and resolution. |
| Gaussian, Sato, and Hessian Filters | Image processing filters used to smooth signals, highlight curvilinear structures, and generate binary images of fibers. |
Protocol Workflow:
- Sample Preparation and Imaging: Culture cells on an appropriate substrate like laminin. Perform immunofluorescence staining using an antibody against α-tubulin to label the microtubule network. Acquire high-resolution images using a fluorescence microscope, capturing multiple Z-slices for each field of view [1].
- Image Pre-processing: Apply deconvolution to the Z-stack images to reduce noise and enhance clarity. Perform a maximum intensity projection (MIP) to create a single 2D composite image from the Z-stack for analysis [1].
- Fiber Enhancement and Segmentation: Process the projected image with a Gaussian filter to smooth the fluorescence signal. Subsequently, apply a Sato filter, which is specifically designed to enhance curvilinear structures, making individual microtubule fibers more distinct. Finally, use a Hessian filter to generate a binary image, separating the fiber structures from the background [1].
- Skeletonization and Feature Extraction: Skeletonize the binary image to reduce each fiber to a single-pixel-wide line. This skeleton is then used for automatic extraction of two classes of features:
- Line Segment Features (LSFs): Treats the skeleton as a collection of line segments to quantify metrics like fiber length, orientation (OOP), and quantity (Nl) [1].
- Cytoskeleton Network Features (CNFs): Represents the skeleton as a graph network of nodes and edges to quantify connectivity, complexity, and radiality relative to the nucleus centroid [1].
- Data Analysis and Validation: Compare the extracted feature sets (e.g., OOP, compactness, radiality) between control (e.g., wild-type E-cadherin) and experimental (e.g., mutant E-cadherin) cell populations to identify statistically significant alterations associated with the invasive phenotype [1].
Directionality Analysis with TeDT
For a more focused analysis of microtubule orientation, the Texture Detection Technique (TeDT) provides a robust solution. This software tool is based on the Haralick texture method and quantifies directionality by considering both local and global image features, with greater weight on the latter [2]. It is particularly useful for complex microtubule patterns that are difficult to assess visually.
Protocol Workflow:
- Input: Use immunofluorescence images of microtubules.
- Analysis: Run the TeDT algorithm, which expresses results in a graphic form that is responsive to subtle variations in microtubule distribution.
- Output: A numerical score that allows for the quantitation of overall directionality, enabling direct comparison between cell states [2].
Visualization of Experimental Workflows
The following diagrams illustrate the core experimental and analytical processes described in this guide.
Computational Analysis Pipeline
Diagram Title: Workflow for Automated Microtubule Feature Extraction
Microtubule Signature Feature Relationships
Diagram Title: Microtubule Signature Relationships in Invasive Cells
The actin cytoskeleton serves as a primary mechanical engine for cell motility, generating the protrusive and contractile forces necessary for cellular movement. In pathological contexts such as cancer metastasis, the dysregulation of actin dynamics is a critical factor driving the transition from a non-invasive to an invasive phenotype. Invasive cells exhibit distinct cytoskeletal architectures characterized by altered filament organization, dynamics, and mechanical properties compared to their non-invasive counterparts. These differences are not merely morphological but are fundamental to the enhanced migratory and force-generating capabilities of invasive cells. This guide provides a comparative analysis of the experimental frameworks and quantitative data used to dissect these differences, offering researchers a detailed overview of methodologies, key regulatory components, and analytical tools for studying actin-based motility.
Experimental Protocols for Analyzing Actin Architecture and Dynamics
Computational Pipeline for Cytoskeletal Feature Extraction
A novel bioimage analysis pipeline enables quantitative dissection of the cytoskeleton's spatial organization from standard immunofluorescence images. This method is particularly suited for comparing invasive and non-invasive cells [1].
- Sample Preparation and Imaging: Cells are cultured on appropriate substrates (e.g., laminin for a supportive environment), fixed, and stained for a cytoskeletal component such as α-tubulin (for microtubules) or phalloidin for F-actin. The nucleus is also stained. Multiple images are acquired for each channel along the Z-axis [1].
- Image Pre-processing: Z-stacks are projected into 2D using maximum intensity projection (MIP). Deconvolution is applied to remove noise and blur, improving contrast and resolution. A Gaussian filter is then used to smooth the fluorescence signal of cytoskeletal components [1].
- Fiber Segmentation and Skeletonization: A Sato filter is applied to highlight curvilinear structures, and a Hessian filter helps generate binary images. The binary images are then skeletonized, reducing the cytoskeletal network to single-pixel-wide lines for quantitative analysis [1].
- Feature Extraction: The skeletonized image is analyzed to extract two classes of features:
- Line Segment Features (LSFs): Describe the morphology and distribution of individual fibers, including parameters like length, orientation, and quantity [1].
- Cytoskeleton Network Features (CNFs): Describe the topological properties of the network using graph theory, quantifying connectivity, complexity, and radiality relative to the nucleus centroid [1].
- Application Note: This pipeline was validated using cells expressing wild-type E-cadherin versus a mutant E-cadherin (p.L13_L15del) that causes loss of cell-cell adhesion and increases invasiveness. The method successfully distinguished unique microtubule signatures between the two cell types [1].
Fluorescence Recovery After Photobleaching (FRAP) for Actin Turnover
FRAP is used to measure the dynamics and turnover of actin filaments, differentiating between populations of stable and dynamic actin [3].
- Cell Preparation: Neurons or other cell types expressing a fluorescently tagged actin (e.g., GFP-Actin) are used. The cells should be in a mature state (e.g., 14 days in vitro for neurons) [3].
- Photobleaching and Recovery: A specific region of interest within a cellular structure, such as a dendritic spine, is bleached using a high-intensity laser beam, permanently disabling the fluorescence of the tagged proteins in that area. The laser is then switched back to a low-intensity setting to monitor the fluorescence recovery over time [3].
- Data Analysis: The recovery kinetics are quantified. The mobile fraction of actin and the half-time of recovery provide information on actin turnover rates. A persistent, non-recovering fraction indicates a stable pool of actin that does not exchange rapidly with the surrounding cytoplasm. This stable pool is often associated with cross-linked filaments [3].
- Application Note: This protocol can reveal long-term changes in actin stability following stimuli. For instance, hours after the induction of chemical Long-Term Potentiation (cLTP), a 2-3 fold increase in the stable actin pool was observed in dendritic spines, which is crucial for maintaining structural changes [3].
Quantitative Comparison of Cytoskeletal Features
The following tables summarize key quantitative differences in cytoskeletal organization and the effects of various perturbations, as identified through the described methodologies.
Table 1: Quantitative Cytoskeletal Features in Invasive vs. Non-Invasive Cells
| Parameter | Description | Non-Invasive Cells (Wild-Type E-cadherin) | Invasive Cells (Mutant E-cadherin) | Measurement Method |
|---|---|---|---|---|
| Orientational Order Parameter (OOP) | Measures fiber alignment; higher value = more aligned fibers. | Higher OOP values | Significantly lower OOP values [1] | Computational Pipeline [1] |
| Fiber Compactness (Nl/Ac) | Number of fibers per unit cell area. | Less compact, more dispersed fibers (e.g., 0.421 μm⁻²) [1] | More compactly distributed fibers (e.g., 1.539-2.039 μm⁻²) [1] | Computational Pipeline [1] |
| Fiber Length Variability | Intercellular variability in the length of fibers. | Lower variability | Higher variability [1] | Computational Pipeline [1] |
| Radiality Score (RS) | Measures how much fibers nucleate from the nucleus centroid. | Variable, but can be low in round cells (e.g., 0.266) [1] | Can show a more prominent radial pattern (e.g., 0.564) [1] | Computational Pipeline [1] |
Table 2: Effects of Genetic and Pharmacological Perturbations on Actin Dynamics
| Perturbation / Condition | Biological Context | Key Observed Effects on Cytoskeleton & Motility | Experimental Model |
|---|---|---|---|
| Cofilin Deficiency | T cell development | Severe early block in thymocyte development; accumulated F-actin, impaired migration and synapse disassembly [4]. | Genetic mouse model (Cfl1 mutation) [4] |
| WASP/ARPC1B Mutation | Immunodeficiency (WAS) | Defective branched actin polymerization; aberrant actin spikes/filopodia; unstable cell conjugates and impaired cytotoxicity [4]. | Patient T cells [4] |
| ATM-3507 (Tpm3.1/3.2 inhibitor) | B-cell Lymphoma (DLBCL) | Disrupted peripheral actin ring and actomyosin arcs; inhibited BCR-induced spreading, growth, and chemotaxis [5]. | DLBCL cell lines [5] |
| Non-Invasive Physical Plasma (NIPP) | Various Cancer Cell Lines | Disrupted cytoskeletal organization; altered metabolic activity; inhibited proliferation and migration [6]. | Ovarian, prostate, and breast cancer cell lines [6] |
| Long-Term Potentiation (LTP) | Neuronal Dendritic Spines | Rapid spine volume increase (~150%); long-term (hours) 2-3 fold increase in the stable, cross-linked actin pool [3]. | Hippocampal cultured neurons [3] |
Key Signaling Pathways and Molecular Mechanisms
Actin Regulators in Protrusion and Motility
The core machinery for generating protrusive forces involves a coordinated system of nucleators, binding proteins, and small GTPases. The Arp2/3 complex, activated by Nucleation-Promoting Factors (NPFs) like WASP and WAVE2, generates branched actin networks that drive lamellipodial protrusions at the leading edge. Simultaneously, formin proteins (e.g., mDia1) generate linear actin filaments and arcs, which cooperate with myosin II to contract and transport cargo inward [4]. The balance between polymerization and depolymerization is fine-tuned by proteins like cofilin, which severs old filaments to replenish the monomer pool [4]. In motile cells such as lymphocytes, this machinery is polarized to form an immunological synapse, with continuous retrograde actin flow corralling receptors and stabilizing contact with target cells [4].
Diagram Title: Actin Regulatory Network for Motility
Force Generation and Mechanosensing by Myosin Filaments
Myosin II is the primary motor protein generating contractile forces. It self-assembles into bipolar filaments that walk toward the barbed ends of actin filaments. The structural properties of these filaments—including the number of myosin heads, the length of the filament, and the size of the central bare zone—directly impact the magnitude and efficiency of force generation [7]. In disorganized actomyosin networks, myosin II filaments generate tensile forces by pulling anti-parallel actin filaments together, while compressive forces are dissipated through filament buckling. Computational models show that cooperative effects between multiple myosin filaments can enhance total force output, and the presence of passive actin cross-linkers can stabilize the network against these forces [7]. This contractile apparatus is essential for processes like cytokinesis, focal adhesion turnover, and the retrograde flow of actin at the immunological synapse.
Actin Repair by Force-Activated Zyxin
The cytoskeleton is subject to mechanical stress and damage. The LIM-domain protein zyxin acts as a force-sensitive sensor that localizes to sites of actin filament rupture. Upon damage, zyxin forms force-dependent assemblies that bridge broken filament fragments. These assemblies then serve as a platform to recruit and coordinate repair factors: they recruit VASP to nucleate new actin filaments and α-actinin to crosslink them into aligned bundles, thereby rapidly restoring the integrity of the stress fiber [8]. This mechanism operates at the network scale to maintain cytoskeletal integrity under mechanical load.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying Actin Dynamics and Motility
| Reagent / Tool | Function / Target | Key Application in Research |
|---|---|---|
| ATM-3507 (Anisina) | Selective inhibitor of Tpm3.1/3.2 tropomyosin isoforms [5]. | Disrupts Tpm3.1/3.2-stabilized actin filaments; used to study roles in B-cell spreading, lymphoma growth, and migration [5]. |
| CRISPR/Cas9 Gene Editing | Targeted gene knockout or mutation. | Generating deficient cell lines (e.g., cofilin, WASP, ARPC1B) to study protein function in actin polymerization and T cell function [4]. |
| Reconstituted Systems | Purified proteins (actin, NPFs, Arp2/3, etc.) assembled in vitro [9]. | Minimal system to study actin network assembly, force generation, and interaction with membranes or beads, free from cellular complexity [9] [8]. |
| Agent-Based Computational Models | In silico simulation of actomyosin structures [7]. | Modeling force generation by myosin II filaments with detailed structural properties in bundles and networks [7]. |
| Non-Invasive Physical Plasma (NIPP) | Induces oxidative stress and metabolic disruption [6]. | Modulates cancer cell cytoskeleton, growth, and motility without inducing cytoprotective heat shock proteins [6]. |
Visualizing the Computational Analysis Workflow
The following diagram outlines the key steps in the computational pipeline for quantifying cytoskeletal architecture, providing a clear workflow for researchers looking to implement this approach.
Diagram Title: Cytoskeletal Analysis Computational Pipeline
The cytoskeleton is a complex, dynamic network critical for maintaining cellular architecture and function. Its three major components—actin filaments, microtubules, and intermediate filaments—do not operate in isolation but are functionally integrated by specialized proteins known as cytoskeletal crosslinkers. Among these, plectin stands out as a versatile and crucial cytolinker that facilitates cytoskeletal crosstalk and mechanotransduction. This review synthesizes current research comparing cytoskeletal organization in invasive and non-invasive cells, with a focused analysis on plectin's role as a master integrator of mechanical signaling. We examine how plectin-mediated networks differ between these cellular states and evaluate emerging therapeutic strategies that target this crosslinking functionality, providing a comparative guide grounded in experimental data.
Plectin: A Master Cytoskeletal Integrator
Plectin is a giant (~500 kDa) cytolinker protein encoded by the PLEC gene, located on chromosome 8 (8q24) [10]. Its structural organization includes an N-terminal actin-binding domain (ABD), a central plakin domain, a rod domain, and a C-terminal intermediate filament-binding domain [10]. This multidomain architecture enables plectin to crosslink all three major cytoskeletal filament systems: actin filaments, intermediate filaments (particularly vimentin), and microtubules [10]. Through alternative splicing of its first exons, plectin generates multiple isoforms that target distinct cellular structures, including plectin 1a (hemidesmosomes), 1b (mitochondria), 1c (microtubules), and 1f (focal adhesions) [10].
Plectin's crosslinking activity is mechanosensitive, responding to actomyosin contractility and substrate stiffness [11]. This property positions plectin as a key regulator of cellular tensional homeostasis, enabling cells to sense and respond to mechanical cues from their extracellular environment [12] [13]. In migrating cells, plectin facilitates the interaction between F-actin and vimentin intermediate filaments in an actomyosin-dependent manner, creating integrated networks that withstand mechanical stress during cell movement [11].
Experimental Approaches for Analyzing Cytoskeletal Organization
Methodologies for Assessing Cytoskeletal Crosstalk
Investigating plectin-mediated cytoskeletal integration requires specialized experimental approaches that can capture both structural organization and dynamic interactions:
Proximity Ligation Assay (PLA): This technique generates fluorescent signals when antibodies bound to target proteins are within 40 nm, allowing precise detection of protein-protein interactions. Researchers have employed PLA to demonstrate plectin-dependent crosslinking between actin and vimentin networks, with significant reduction in PLA puncta observed following plectin knockdown [11].
Intermolecular FRET Imaging: Fluorescence resonance energy transfer (FRET) between fluorophore-tagged cytoskeletal components (e.g., mTurquoise-actin and mNeongreen-vimentin) confirms their close molecular interactions in live cells migrating through 3D environments. This approach has revealed that actin-vimentin interactions are partially dependent on ROCK-mediated actomyosin contractility [11].
Computational Cytoskeletal Analysis: Novel image-based pipelines enable quantitative characterization of cytoskeletal architecture from immunofluorescence images. This methodology involves deconvolution, Gaussian and Sato filtering, Hessian analysis, and skeletonization to extract parameters including fiber orientation, morphology, compactness, and radiality relative to the nucleus [1].
Genetic and Pharmacological Perturbation: Plectin function can be disrupted using siRNA-mediated knockdown (achieving ~62% reduction in expression) [11], CRISPR/Cas9-generated knockout cells [12] [13], and the ruthenium-based inhibitor plecstatin-1 (PST) [12] [13]. These approaches enable comparative analysis of cytoskeletal organization and cell behavior with and without functional plectin.
Workflow for Cytoskeletal Architecture Analysis
The following diagram illustrates the integrated experimental and computational workflow for analyzing cytoskeletal organization in invasive and non-invasive cells:
Comparative Cytoskeletal Architecture: Invasive vs. Non-Invasive Cells
Quantitative Differences in Cytoskeletal Organization
Computational analysis of cytoskeletal architecture reveals distinct organizational patterns between invasive and non-invasive cells. The table below summarizes key differences identified through quantitative imaging:
Table 1: Cytoskeletal Features in Invasive Versus Non-Invasive Cells
| Cytoskeletal Feature | Non-Invasive Cells | Invasive Cells | Measurement Method |
|---|---|---|---|
| Microtubule Orientation | Higher OOP values (0.475) indicating aligned fibers | Lower OOP values (0.019) indicating disorganized fibers | Orientational Order Parameter (OOP) [1] |
| Microtubule Length | Variable | Shorter fibers with higher length variability | Line Segment Extraction (LiE) [1] |
| Fiber Compactness | More dispersed distribution (0.421 μm⁻²) | More compact distribution (2.039 μm⁻²) | Fibers per cell area (Nl/Ac) [1] |
| Radial Organization | Lower radial scores (0.266) | Higher radial scores (0.564) | Radiality Score (RS) [1] |
| Fiber-Nucleus Distance | Shorter distances (3.14 μm) | Longer distances (15.94 μm) | Distance to nucleus centroid (Di) [1] |
| Actin-Vimentin Interaction | Plectin-dependent, mechanosensitive | Enhanced, contractility-dependent | Proximity Ligation Assay (PLA) [11] |
Plectin Expression and Localization Patterns
Plectin demonstrates distinct expression and subcellular localization patterns that correlate with invasive potential:
Table 2: Plectin Dysregulation in Cancer Cells
| Aspect of Dysregulation | Non-Invasive/Well-Differentiated Cells | Invasive/Poorly-Differentiated Cells | Functional Consequences |
|---|---|---|---|
| Expression Level | Lower plectin expression | Elevated plectin levels (HCC, pancreatic, ovarian cancer) [12] [10] | Promoted migration, invasion, and metastasis [12] [13] |
| Subcellular Localization | Cytoplasmic distribution | Perimembranous enrichment; cell surface presentation (CSP) [12] [10] | Enhanced mechanosignaling; novel functions in migration [12] [10] |
| Isoform Expression | Balanced isoform profile | Isoform-specific dysregulation (e.g., plectin 1f in focal adhesions) [10] | Targeted cytoskeletal remodeling at specific locations [10] |
| Prognostic Value | Not applicable | Correlates with poor survival and decreased recurrence-free survival in HCC [12] [13] | Potential as diagnostic and prognostic biomarker [12] [13] |
Plectin in Mechanosignaling and 3D Cell Migration
Mechanisms of Plectin-Mediated Mechanotransduction
In invasive cells, plectin integrates mechanical signals from the extracellular matrix through focal adhesions, activating key oncogenic signaling pathways. The following diagram illustrates plectin's role in mechanotransduction:
Plectin inactivation through genetic ablation or pharmacological inhibition with plecstatin-1 disrupts this mechanical signaling cascade, attenuating FAK, MAPK/Erk, and PI3K/AKT pathway activation and consequently suppressing tumor growth and metastasis in hepatocellular carcinoma models [12] [13] [14].
Role in 3D Migration and Nuclear Piston Mechanism
Plectin facilitates a "nuclear piston" migration mechanism essential for invasive movement through 3D matrices. This process involves:
Cytoskeletal Polarization: Plectin crosslinks vimentin and actomyosin filaments, localizing the vimentin network around the nucleus and polarizing non-muscle myosin II (NMII) anterior to the nucleus [11].
Nuclear Translocation: Plectin-mediated connections between the vimentin network and actomyosin filaments enable forward pulling of the nucleus as cells migrate through confined 3D spaces [11].
Pressure Compartmentalization: The nucleus acts as a piston, generating compartmentalized intracellular pressure with high-pressure anterior cytoplasmic compartments and lower-pressure rear compartments, facilitating lobopodial protrusions [11].
Mechanosensitive Regulation: Plectin-vimentin interactions are mechanosensitive, responding to NMII contractility and substrate stiffness to activate the nuclear piston machinery specifically in cross-linked 3D environments [11].
Plectin knockdown disrupts these processes, impairing 3D migration without significantly affecting 2D movement, highlighting its specific role in invasive migration through complex microenvironments [11].
Therapeutic Targeting of Plectin-Mediated Mechanisms
Research Reagent Solutions for Plectin Investigation
Table 3: Essential Research Tools for Studying Plectin Function
| Reagent/Technique | Specific Example | Research Application | Experimental Outcome |
|---|---|---|---|
| siRNA Knockdown | Pan-plectin siRNA | Reduces plectin expression by ~62% [11] | Decreased actin-vimentin PLA puncta; impaired 3D migration [11] |
| Pharmacological Inhibitor | Plecstatin-1 (PST) | Ruthenium-based plectin inhibitor [12] [13] | Suppressed HCC growth, invasion, and metastasis in mouse models [12] [13] |
| CRISPR/Cas9 Knockout | PLEC-deficient HCC cells | Complete genetic ablation of plectin [12] [13] | Limited migration, invasion, and anchorage-independent growth [12] [13] |
| Proximity Ligation Assay | Actin-Vimentin PLA | Detects <40 nm interactions between cytoskeletal networks [11] | Quantified plectin-dependent cytoskeletal crosslinking [11] |
| FRET Imaging | mTurquoise-actin + mNeongreen-vimentin | Live-cell monitoring of cytoskeletal interactions [11] | Confirmed actomyosin-dependent actin-vimentin association [11] |
Emerging Therapeutic Applications
Targeting plectin-mediated cytoskeletal integration represents a promising therapeutic strategy for invasive cancers:
Plecstatin-1 in HCC: Treatment with PST inhibited hepatocellular carcinoma initiation and growth in autochthonous and orthotopic mouse models, reducing metastatic outgrowth in lungs by disrupting oncogenic mechanosignaling [12] [13] [14].
Non-Invasive Physical Plasma (NIPP): This novel approach disrupts cytoskeletal organization across ovarian, prostate, and breast cancer cell lines by inducing oxidative stress that damages actin and tubulin without triggering cytoprotective heat shock proteins [6] [15].
Antibody-Based Therapies: Monoclonal antibodies targeting cancer-specific plectin (CSP) on the cell surface show potential for selective drug delivery in ovarian cancer [10].
These approaches demonstrate the therapeutic potential of disrupting plectin-mediated cytoskeletal integration to combat invasive cancers, with plecstatin-1 showing particular promise in preclinical HCC models [12] [13].
Plectin serves as a critical integrator of cytoskeletal networks, with distinct expression patterns, organizational roles, and functional contributions that differentiate invasive from non-invasive cells. Through its mechanosensitive crosslinking capabilities, plectin regulates essential processes in cell invasion, including 3D migration, nuclear translocation, and oncogenic mechanosignaling. The comparative analysis presented herein provides researchers with methodological frameworks, quantitative benchmarks, and therapeutic insights for investigating cytoskeletal organization in the context of cell invasiveness. Emerging strategies that target plectin-mediated mechanisms offer promising avenues for therapeutic intervention in invasive cancers, particularly through the disruption of mechanical homeostasis that drives tumor progression and metastasis.
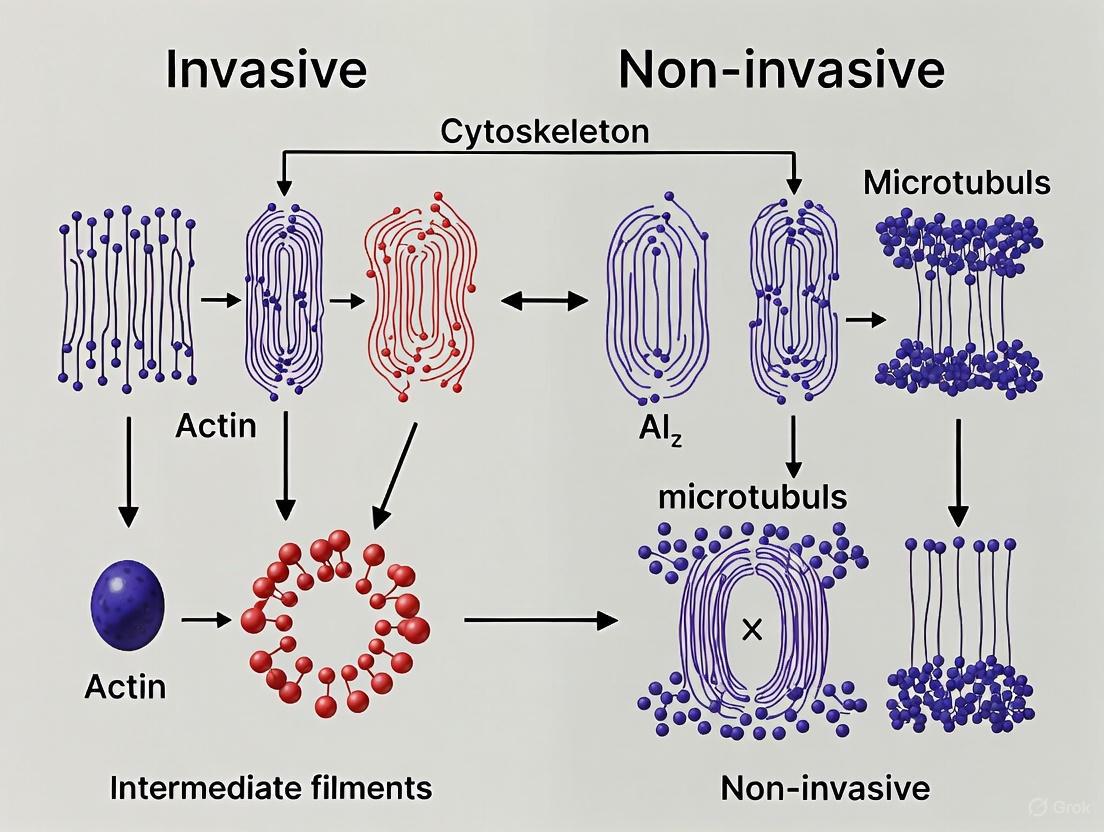
E-Cadherin Disruption and its Cascading Effects on Cytoskeletal Architecture
| Feature | Wild-Type (Non-Invasive) E-Cadherin Cells | Mutant (Invasive) E-Cadherin Cells |
|---|---|---|
| Microtubule Organization (OOP) [1] | Higher Orientational Order Parameter (OOP) | Significantly lower OOP |
| Microtubule Morphology [1] | Longer fibers | Shorter fibers |
| Microtubule Distribution [1] | More dispersed in the cytoplasm | More compactly distributed |
| Microtubule Radiality [1] | Variable radial patterns | Dispersed orientations, less radial |
| Primary Extrusion Direction [16] | Primarily apical extrusion | Preferential basal extrusion into the ECM |
| Cell-ECM Adhesion [16] | Normal adhesion strength | Increased adhesion to ECM |
| Invadopodia Formation [17] | Not typically observed | E-cadherin localizes to invadopodia, promoting structuration and ECM degradation |
Experimental Workflow for Cytoskeletal Analysis
A novel computational pipeline enables a detailed quantitative comparison of cytoskeletal architecture between non-invasive and invasive cells [1]. The methodology below allows researchers to systematically dissect the subtle cytoskeletal alterations triggered by E-cadherin disruption.
Detailed Experimental Protocols
This protocol is designed to quantify fine alterations in the cytoskeleton of cells where E-cadherin function has been disrupted.
- Cell Culture and Transfection: Culture control cells (e.g., MDCK or MCF10A) and isogenic lines expressing mutant E-cadherin (e.g., p.L13_L15del). Grow cells on appropriate ECM-coated glass-bottom dishes to model cell-ECM interaction.
- Immunofluorescence Staining:
- Fix cells with 4% paraformaldehyde for 15 minutes.
- Permeabilize with 0.1% Triton X-100 for 10 minutes.
- Block with 1% BSA for 1 hour.
- Incubate with primary antibody against α-tubulin (microtubule marker) overnight at 4°C.
- Incubate with fluorescent secondary antibody (e.g., Alexa Fluor 488) for 1 hour at room temperature.
- Counterstain nuclei with DAPI.
- Image Acquisition: Acquire high-resolution z-stack images using a confocal microscope with a 60x or higher magnification oil-immersion objective. Maintain identical laser power and gain settings across all samples.
- Image Processing and Analysis:
- Pre-processing: Apply deconvolution to remove noise and blur. Create a 2D maximum intensity projection (MIP) of the z-stacks.
- Filtering: Process images with a Gaussian filter to smooth the signal, followed by a Sato filter to highlight curvilinear structures of cytoskeletal fibers.
- Segmentation: Apply a Hessian filter to generate binary images. Skeletonize the binary images to create a 1-pixel-wide representation of each fiber.
- Feature Extraction: Use custom algorithms to automatically extract two classes of features:
- Line Segment Features (LSFs): Quantify fiber length, orientation (Orientational Order Parameter - OOP), and quantity (number of lines, Nl).
- Cytoskeleton Network Features (CNFs): Analyze compactness (Nl/Ac), radiality (radial score relative to nucleus centroid), and fiber-nucleus interconnection (average distance, Di).
This assay tests the ability of E-cadherin dysfunctional cells to detach from an epithelium and invade the underlying matrix, mimicking an early step in cancer dissemination.
- Generation of Co-culture Model:
- Label E-cadherin mutant cells (e.g., A634V, R749W, V832M) with a fluorescent cell tracker dye.
- Mix these labeled mutant cells at a highly diluted ratio (e.g., 1:100) with an excess of unlabeled wild-type cells.
- Seed the cell mixture on top of a thick collagen I matrix to form a confluent monolayer.
- Confocal Microscopy and Quantification:
- After 24-48 hours of culture, acquire xz-sections (side views) of the monolayer using confocal microscopy.
- Quantify the position of the nuclei of mutant (fluorescent) and wild-type cells relative to the apical-basal axis of the monolayer.
- A cell is scored as "basally extruded" if its nucleus is located below the basal plane of the wild-type monolayer.
E-Cadherin Disruption and Downstream Signaling
E-cadherin is not merely a passive adhesive molecule; its engagement or disruption initiates powerful signaling cascades that directly remodel the actin cytoskeleton, primarily through the Rho family of GTPases [18].
The Scientist's Toolkit: Key Research Reagents
| Research Reagent / Material | Function in Experiment | Example Application |
|---|---|---|
| Mutant E-cadherin Constructs | Model hereditary E-cadherin dysfunction to study loss of cell-cell adhesion. | p.L13_L15del, A634V, R749W, V832M mutants in HDGC research [1] [16]. |
| Laminin / Collagen I Matrix | Provides a physiological, supportive 3D environment for cell growth and invasion assays. | Substrate for cell culture in cytoskeleton analysis and basal extrusion assays [1] [16]. |
| Anti-α-Tubulin Antibody | Primary antibody for visualizing the microtubule network via immunofluorescence. | Key reagent for staining and segmenting cytoskeletal fibers [1]. |
| Pefabloc (Serine Protease Inhibitor) | Inhibits serine proteases like FAP/seprase to test their role in invasion. | Validates role of FAP in cancer cell invasion through urothelial barrier [19]. |
| Fluorescent Cell Tracker Dyes | Labels specific cell populations for tracking their behavior in co-culture. | Distinguishes E-cadherin mutant cells from wild-type neighbors in extrusion assays [16]. |
| CRISPR-Cas9 Engineered Cells | Creates isogenic cell lines with precise knockouts (e.g., CDH1+/-) to study E-cadherin loss. | Modeling E-cadherin downregulation in airway epithelial barrier dysfunction [20]. |
The quantitative data and experimental workflows presented provide a robust framework for objectively comparing cytoskeletal organization in invasive versus non-invasive cellular models. The integration of computational image analysis with mechanistic biochemical assays allows for a comprehensive dissection of how E-cadherin disruption serves as a master regulator of cytoskeletal dynamics, driving pro-invasive cellular phenotypes.
{}
Cytoskeletal Drugs as Probes: Unraveling Function Through Targeted Disruption
The cytoskeleton, a dynamic network of filamentous proteins, is a central regulator of cellular architecture, mechanics, and motility. Its dysregulation is a hallmark of numerous diseases, particularly cancer, where it drives invasion and metastasis. This guide provides a comparative analysis of cytoskeleton-targeting drugs, evaluating their efficacy as biological probes and their potential as therapeutics. We objectively compare the performance of actin filament disruptors, Rho kinase inhibitors, and microtubule-targeting agents, supported by experimental data on their morphological, mechanical, and functional impacts on cells. Framed within the context of cytoskeletal organization in invasive versus non-invasive cells, this review integrates detailed experimental protocols, visualizes key signaling pathways, and catalogs essential research reagents to serve as a comprehensive resource for researchers and drug development professionals.
The cytoskeleton is a complex, integrated system composed of three primary filament types: actin filaments (AFs), microtubules (MTs), and intermediate filaments (IFs). This network is not a static scaffold but a dynamic structure that governs essential processes such as cell division, migration, and intracellular transport [21] [22]. In cancer, the cytoskeleton is hijacked to enable invasive behaviors; cells undergo dramatic rearrangements of their cytoskeletal architecture to breach tissues, enter the circulation, and form metastases [1] [22]. Consequently, drugs that selectively disrupt specific cytoskeletal components have become indispensable tools for dissecting the contribution of each filament system to cellular function and pathology.
These "cytoskeletal drugs" function through distinct mechanisms. Actin filament disruptors, such as Latrunculin A, directly target the polymerization dynamics of actin. In contrast, Rho kinase (ROCK) inhibitors act upstream, modulating the signaling pathways that control actomyosin contractility [23]. The specific disruption caused by these drugs allows researchers to infer the normal function of the cytoskeletal components they target. By comparing the effects of these agents on cellular morphology, mechanical properties, and migratory behavior—particularly in models of invasive and non-invasive cells—we can unravel the fundamental mechanisms driving diseases like cancer and identify potential therapeutic vulnerabilities.
Comparative Analysis of Major Cytoskeletal Drug Classes
This section provides a detailed, data-driven comparison of the primary classes of cytoskeleton-modulating agents, focusing on their mechanisms, cellular effects, and experimental outcomes.
Actin Filament Disruptors
Actin filament disruptors directly interfere with the polymerization or stability of actin, leading to a rapid dissolution of actin-based structures. The table below summarizes key drugs in this class and their observed experimental effects.
Table 1: Comparison of Actin Filament Disruptors
| Drug Name | Primary Mechanism of Action | Effect on Actin Structures | Quantified Effect on Cell Elasticity | Impact on Cell Migration/Invasion |
|---|---|---|---|---|
| Latrunculin A | Sequesters G-actin monomers, preventing polymerization [23]. | Disassembles stress fibers and cortical actin [23]. | Distinct decrease in elastic modulus [24]. | Significantly inhibits migration and invasion [6]. |
| Cytochalasin D | Caps filament ends, preventing elongation [23]. | Disassembles stress fibers; can cause actin aggregation [24]. | Distinct decrease in elastic modulus [24]. | Inhibits migration by disrupting leading edge protrusions. |
| Jasplakinolide | Stabilizes actin filaments and promotes polymerization [24]. | Disaggregates filaments but does not disassemble stress fibers [24]. | No significant change in elastic modulus [24]. | Can inhibit migration by reducing filament turnover. |
Rho Kinase (ROCK) Inhibitors
ROCK inhibitors act by disrupting the Rho/ROCK signaling pathway, which is a major regulator of actomyosin contractility. By inhibiting myosin light chain phosphorylation, they indirectly cause actin cytoskeleton reorganization.
Table 2: Comparison of Rho Kinase (ROCK) Inhibitors
| Drug Name | Primary Mechanism of Action | Effect on Actin Structures & Contractility | Documented Effect on Tissue/Outflow | Key Experimental Findings |
|---|---|---|---|---|
| Y-27632 | Specific inhibitor of Rho kinase [23]. | Reduces MLC phosphorylation, leading to stress fiber disassembly and loss of focal adhesions [23]. | Relaxes contractions in TM strips; decreases outflow resistance in animal models [23]. | Induces cell rounding and intercellular separation; mimics effects of direct actin disruption. |
| H-1152 | Potent and selective ROCK inhibitor [23]. | Reduces basal MLC phosphorylation, changing cell shape and depolymerizing actin [23]. | Effectively decreases intraocular pressure in models [23]. | More potent than Y-27632 in some physiological assays. |
Microtubule-Targeting Agents
While the provided search results focus more on actin-targeted therapies, microtubules are a co-conspirator in cancer progression. The computational pipeline study revealed that microtubules in invasive cells with disrupted E-cadherin are shorter, have dispersed orientations, and are more compactly distributed [1]. This suggests that drugs stabilizing or destabilizing microtubules (e.g., Taxol, Vinca alkaloids) would profoundly alter the invasive architecture of cancer cells, though their use as probes is complicated by their essential role in mitosis.
Experimental Protocols for Probing Cytoskeletal Function
To ensure reproducibility and provide a clear framework for comparison, this section outlines standard methodologies for assessing the effects of cytoskeletal drugs.
Protocol: Quantifying Drug-Induced Changes in Cell Mechanics via Atomic Force Microscopy (AFM)
This protocol is adapted from studies investigating the mechanical role of the cytoskeleton [24].
- Cell Seeding: Plate cells (e.g., fibroblasts or cancer cells) onto sterile, glass-bottom culture dishes and allow them to adhere for 24-48 hours until they reach 60-70% confluence.
- Drug Treatment: Replace the medium with a serum-free medium containing the desired concentration of cytoskeletal drug (e.g., 1 µM Latrunculin A, 10 µM Cytochalasin D, or 10 µM Y-27632). Include a vehicle control (e.g., DMSO). Incubate for the determined time (e.g., 30-60 minutes).
- AFM Measurement:
- Use an AFM equipped with a colloidal probe (e.g., a silica bead with a 5-10 µm diameter) to ensure non-indenting measurements.
- Approach the cell surface at multiple locations (avoiding the nucleus) to obtain force-indentation curves.
- Maintain a consistent culture medium and temperature (37°C) during measurements.
- Data Analysis: Fit the force-indentation curves with an appropriate mechanical model (e.g., Hertz model) to extract the Young's modulus (Elastic Modulus). Compare the mean values from drug-treated cells to vehicle-treated controls using statistical tests (e.g., t-test). A significant decrease indicates a loss of mechanical stiffness, as seen with actin disruption [24].
Protocol: Assessing Cytoskeletal Architecture via Computational Image Analysis
This workflow, based on a novel computational pipeline, allows for the quantitative dissection of cytoskeletal organization from fluorescence images [1].
- Sample Preparation and Imaging:
- Culture cells on appropriate substrates (e.g., laminin to mimic ECM interaction).
- Fix, permeabilize, and stain cells for a cytoskeletal target (e.g., α-tubulin for microtubules) and the nucleus.
- Acquire high-resolution Z-stack images using a fluorescence microscope.
- Image Pre-processing:
- Apply deconvolution to remove noise and blur.
- Create a 2D maximum intensity projection (MIP) of the Z-stacks.
- Process images with a Gaussian filter to smooth signal and a Sato filter to highlight curvilinear structures.
- Feature Extraction:
- Generate binary images using a Hessian filter and skeletonize the structures.
- Use the pipeline to automatically extract two classes of features:
- Line Segment Features (LSFs): Quantify fiber orientation, length, and compactness.
- Cytoskeleton Network Features (CNFs): Describe connectivity, radiality, and complexity via graph networks.
- Data Interpretation:
- Calculate the Orientational Order Parameter (OOP). A lower OOP indicates disorganized fibers, a signature of invasive cells [1].
- Compare metrics like fiber compactness and radiality between experimental groups (e.g., wild-type vs. mutant E-cadherin cells) to statistically evaluate cytoskeletal reorganization.
Visualizing Signaling Pathways and Experimental Workflows
The following diagrams, generated using Graphviz DOT language, illustrate the core mechanisms and methodologies discussed in this guide.
Rho/ROCK Signaling Pathway in Cytoskeletal Regulation
Diagram 1: Key pathways regulating actin and myosin. This diagram illustrates the Rho/ROCK signaling pathway that regulates actomyosin contractility and stress fiber formation. It highlights the points of inhibition for ROCK inhibitors (like Y-27632) and the direct target of actin filament disruptors (like Latrunculin A).
Computational Image Analysis Pipeline
Diagram 2: Workflow for cytoskeleton architecture analysis. This flowchart outlines the computational pipeline for quantitatively analyzing cytoskeletal architecture from fluorescence images, culminating in the extraction of Line Segment and Cytoskeleton Network Features [1].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogs essential reagents, drugs, and tools for conducting research in cytoskeletal dynamics and targeted disruption.
Table 3: Essential Reagents for Cytoskeletal Disruption Research
| Reagent / Solution | Function / Application | Key Characteristics / Notes |
|---|---|---|
| Latrunculin A & B | Selective actin monomer sequestering agent; disrupts actin dynamics [23] [25]. | Ideal for probing the specific role of actin polymerization in processes like migration and mechanotransduction. |
| Cytochalasin D | Actin filament capping agent; prevents filament elongation [23] [24]. | Useful for rapid disruption of existing actin networks. Can cause actin aggregation. |
| Y-27632 | Cell-permeable, potent ROCK inhibitor [23]. | A standard tool for investigating Rho/ROCK-mediated contractility and its role in cell morphology and tissue function. |
| Jasplakinolide | Actin-stabilizing agent; promotes polymerization [24]. | Used to study the effects of reduced actin turnover. Notably, it does not soften cells in AFM studies [24]. |
| Anti-β-Actin Antibody | Immunofluorescence staining; functional inhibition [25]. | Beyond imaging, specific antibodies can be microinjected to inhibit actin function and disrupt drug resistance [25]. |
| PSC833 & Probenecid | Inhibitors of drug transporters P-glycoprotein and MRP1 [25]. | Used to study the link between cytoskeleton-associated drug resistance and chemotherapeutic efficacy. |
| Laminin-coated Substrata | ECM substrate for cell culture. | Provides a supportive environment for studying cell-ECM interactions and their effect on cytoskeletal organization [1]. |
The targeted disruption of the cytoskeleton with specific pharmacological probes remains a powerful strategy for elucidating its complex functions in health and disease. As this guide has detailed, actin disruptors and ROCK inhibitors are highly effective at breaking down the mechanical integrity and contractile machinery of cells, providing direct evidence for the cytoskeleton's role in processes from glaucoma to cancer metastasis. The experimental data clearly shows that while both classes induce similar phenotypic outcomes like stress fiber loss, their mechanisms—direct molecular intervention versus indirect signaling modulation—are distinct and must be chosen based on the specific research question.
Future research will be shaped by several key frontiers. First, the development of more cell-type-specific agents and prodrugs is crucial to minimize off-target effects and enhance therapeutic potential, as initially explored in glaucoma research [23]. Second, the integration of advanced computational pipelines, like the one described here, will move the field beyond qualitative description to robust, quantitative analysis of cytoskeletal architecture, enabling the discovery of novel, disease-specific signatures [1]. Finally, the emergence of nanomaterials as cytoskeleton-modulating platforms offers exciting possibilities for spatiotemporally controlled drug delivery, potentially overcoming the limitations of conventional small molecules [21]. By combining precise pharmacological probes with quantitative readouts and advanced delivery systems, researchers can continue to unravel the intricate functions of the cytoskeleton and translate these insights into next-generation therapeutics.
From Images to Insights: Cutting-Edge Computational and Real-Time Analysis Tools
The quantitative analysis of cytoskeletal architecture provides critical insights into cellular behavior, particularly in cancer research where specific cytoskeletal patterns are associated with invasive potential [1]. Computational pipelines for automated feature extraction are indispensable for objectively identifying these subtle, yet biologically significant, morphological changes. These tools enable researchers to move beyond qualitative descriptions and extract high-dimensional data on features like line segments and network topology from complex biological images.
This guide compares automated feature extraction pipelines, focusing on their application in quantifying cytoskeletal organization differences between invasive and non-invasive cells. We evaluate computational tools based on their feature extraction capabilities, performance metrics, and applicability to cytoskeletal analysis, providing researchers with objective data to select appropriate methodologies for their specific research contexts.
Comparative Analysis of Feature Extraction Tools
Different computational approaches have been developed for feature extraction from biological images, each with unique strengths in handling specific data types and analytical tasks.
Table 1: Computational Pipelines for Feature Extraction
| Pipeline Name | Primary Function | Segmentation Approach | Tracking Capability | Dimensionality | Key Cytoskeletal Features |
|---|---|---|---|---|---|
| Nellie [26] | Organelle segmentation & tracking | Multiscale Frangi filter, hierarchical deconstruction | Radius-adaptive pattern matching, subvoxel tracking | 2D, 3D, live-cell | Morphology, motility, sub-organellar regions, graph networks |
| Cytoskeletal Pipeline [1] | Cytoskeletal architecture analysis | Gaussian, Sato, & Hessian filtering, skeletonization | Not specified | 2D (from 3D Z-stacks) | Line orientation (OOP), compactness, radiality, fiber length, bundling |
| Geometric Feature Method [27] | 3D line segment extraction | Region growing, geometric feature enhancement, CNN | Not specified | 3D large-scale | Line segment completeness, correctness, hierarchical topology |
Performance and Experimental Data
Performance benchmarking reveals how effectively these pipelines extract meaningful biological data. The following table summarizes key quantitative findings from experimental validations.
Table 2: Experimental Performance Metrics
| Pipeline / Study | Experimental Context | Key Quantitative Findings | Performance Metrics |
|---|---|---|---|
| Cytoskeletal Analysis Pipeline [1] | E-cadherin mutant (invasive) vs. wild-type (non-invasive) cells | Invasive cells: ↓OOP (dispersed orientations), ↓Fiber Length, ↑Compactness [1] | Successfully distinguished invasive vs. non-invasive cytoskeletal architectures |
| Geometric Line Extraction [27] | Large-scale outdoor point clouds (Semantic3D, WHU-TLS) | Completeness: ~86%, Correctness: ~86%, Speed: 25,000 points/sec [27] | High accuracy and efficiency in complex environments |
| Nellie [26] | Diverse organelle segmentation & tracking tasks | Outperformed state-of-the-art tools in segmentation across simulated datasets [26] | Superior to custom-trained Swin UNETR models in generalization [26] |
Detailed Experimental Protocols
Protocol 1: Cytoskeletal Feature Extraction from Immunofluorescence Images
This protocol is designed for quantifying microtubule reorganization in 2D cell cultures, suitable for comparing invasive and non-invasive cancer cell lines [1].
Sample Preparation
- Cell Culture: Plate cells on laminin-coated coverslips to model cell-ECM interactions.
- Staining: Immunofluorescence staining for α-tubulin (microtubules) and a nuclear marker (e.g., DAPI).
- Imaging: Acquire high-resolution Z-stack images using a fluorescence microscope.
Image Preprocessing
- Deconvolution: Remove noise and blur, improving contrast and resolution.
- Z-Stack Projection: Create a 2D maximum intensity projection (MIP) from the deconvoluted Z-stacks.
- Filtering Pipeline:
- Apply a Gaussian filter to smooth the fluorescence signal.
- Apply a Sato filter to highlight curvilinear structures of cytoskeletal fibers.
- Apply a Hessian filter to generate a binary image for segmentation.
Segmentation and Skeletonization
- Process the binary image to create a skeleton representation of the cytoskeletal network.
Feature Extraction
- Line Segment Features (LSFs): Extract features like length and orientation from the skeleton.
- Cytoskeleton Network Features (CNFs): Calculate graph-based features from network nodes.
- Key Metrics:
- Orientational Order Parameter (OOP): Measures fiber alignment. Lower angular distribution yields higher OOP.
- Fiber Compactness: Number of fibers per unit area (Nl/Ac).
- Radiality Score (RS): Measures how fibers radiate from the nucleus centroid.
The workflow for this analysis is summarized in the following diagram:
Protocol 2: Nellie for 3D Organelle Segmentation and Tracking
This protocol is used for 3D analysis of organelle morphology and dynamics, which can be applied to study cytoskeletal components in volumetric data [26].
Input Data Preparation
- Acquire 2D/3D or 4D (3D + time) live-cell microscopy images.
- Ensure proper image metadata (voxel dimensions, time interval) is available.
Metadata Validation and Preprocessing
- Metadata Module: Nellie automatically detects dimension order and resolutions, allowing for user correction.
- Multiscale Filtering: A modified Frangi filter enhances structural contrast based on local architecture, not just intensity, adapting to various magnifications.
Hierarchical Segmentation
- Semantic Segmentation: Minotri thresholding creates an initial organellar landscape mask.
- Instance Segmentation: Connected-components labeling identifies spatially disconnected organelles.
- Subcompartments: Skeletonization and junction node detection deconstruct networks into individual branches.
Motion Tracking and Feature Extraction
- Motion-Capture Markers: Generate tracking points independently of segmentation labels for consistency across frames.
- Pattern Matching: Use radius-adaptive, variable-range feature matching to create linkages between time points.
- Feature Output: Extract a hierarchical pool of spatial and temporal features for analysis.
The workflow for this protocol is as follows:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Cytoskeletal Analysis
| Item Name | Function / Application | Relevance to Feature Extraction |
|---|---|---|
| α-Tubulin Antibody [1] | Immunofluorescence staining of microtubules | Primary target for visualizing microtubule networks in the cytoskeletal pipeline. |
| AF488-phalloidin / SiR-jasplakinolide [28] | F-actin staining for fixed / living cells | Labels actin filaments for organization analysis; used in polarimetry. |
| Genetically Encoded Actin Reporters [28] | Live-cell imaging of actin organization (e.g., constrained GFP fusions) | Enables measurement of filament orientation (ρ) and alignment (ψ) via polarimetry in living samples. |
| Laminin-Coated Substrata [1] | Provides physiologically relevant ECM for cell culture | Models cell-ECM interactions crucial for studying invasive phenotypes. |
| Nellie Software [26] | Automated segmentation and tracking pipeline | Provides objective, high-throughput analysis of organelle/cytoskeletal morphology and dynamics. |
| Napari Viewer [26] | User-friendly GUI for image analysis | Facilitates visualization of intermediate images and tracks from Nellie without coding. |
The comparative data indicates that tool selection should be driven by specific research questions. For dedicated 2D cytoskeletal architecture analysis, the specialized pipeline [1] offers targeted metrics like OOP and radiality that directly discriminate invasive phenotypes. For comprehensive 3D and dynamic studies, Nellie's [26] generalizable, hierarchical approach provides unparalleled depth in quantifying morphology and motility.
The consistent finding that invasive cells display shorter microtubules with disorganized orientations and compact distribution [1] provides a quantifiable signature of metastatic potential. Integrating these computational pipelines with advanced imaging techniques, such as polarimetry [28], will further deepen our understanding of cytoskeletal dynamics. These automated, unbiased extraction tools are paving the way for new diagnostic and therapeutic strategies in cancer research.
The architectural organization of cellular and subcellular components is a critical determinant of biological function, particularly in the context of disease progression such as cancer invasion and metastasis. Quantitative parameters that can objectively measure organizational properties provide powerful tools for distinguishing between physiological states that may appear similar through qualitative assessment alone. This guide focuses on three key quantitative parameters—orientational order, fractal dimension, and connectivity—that have emerged as essential metrics for characterizing biological organization, particularly in cytoskeletal architecture and cellular patterning. These parameters enable researchers to move beyond subjective descriptions to objective, computationally-derived measurements that can detect subtle yet biologically significant changes in cellular organization associated with transformative processes like the epithelial-to-mesenchymal transition in cancer cells.
The comparative analysis presented here examines the underlying principles, methodological approaches, and experimental applications of these parameters specifically within the context of distinguishing invasive from non-invasive cellular phenotypes. By providing a structured comparison of these quantitative tools, along with detailed experimental protocols and visualization approaches, this guide aims to equip researchers with the knowledge needed to select and implement the most appropriate parameters for their specific research questions in cytoskeletal organization and cancer cell behavior.
Comparative Analysis of Quantitative Parameters
Table 1: Core Quantitative Parameters for Cellular Organization Analysis
| Parameter | Mathematical Basis | Measurement Range | Biological Interpretation | Invasive vs. Non-Invasive Signatures |
|---|---|---|---|---|
| Orientational Order Parameter (OOP) | Liquid crystal physics; tensor analysis of vector orientation [29] [30] | 0 (isotropic) to 1 (perfectly aligned) | Degree of alignment and directional consistency of cytoskeletal elements | Invasive cells show significantly lower OOP, indicating disorganized fibers [1] |
| Co-Orientational Order Parameter (COOP) | Extension of OOP framework to correlation between two constructs [29] [30] | 0 (uncorrelated) to 1 (perfectly correlated) | Correlation in orientation between different cytoskeletal components (e.g., actin & Z-lines) | Not specifically reported for invasion, but quantifies coordination between structural elements |
| Fractal Dimension (FD) | Modified Blanket Method analyzing power law relationship between surface area and resolution [31] | 2.0-4.5 for biological structures | Structural complexity and space-filling characteristics | Higher FD associates with increased aggressiveness in prostate cancer nuclei [32] |
| Local Connected Fractal Dimension (LCFD) | Local implementation of FD measuring connectivity [32] | Varies by biological context | Regional complexity and interconnection density | LCFD >1.7051 associates with high-aggressiveness prostate carcinomas [32] |
Table 2: Complementary Metrics for Comprehensive Organizational Profiling
| Metric Category | Specific Parameters | Application Context | Invasion-Associated Patterns |
|---|---|---|---|
| Spatial Distribution | Lacunarity (λ) [32] | Measures heterogeneity and "gappiness" of spatial patterns | λ < 0.7 in high-aggression prostate cancers [32] |
| Information Theory | Shannon Entropy (H) [32] | Quantifies disorder and uncertainty in spatial organization | H > 0.9 in high-complexity, aggressive carcinomas [32] |
| Morphometric | Line Segment Features (LSF) and Cytoskeleton Network Features (CNF) [1] | Quantifies fiber length, bundling, and network architecture | Invasive cells show shorter microtubules with dispersed orientations [1] |
| Radiality | Radial Score (RS) [1] | Measures pattern organization relative to a central point (e.g., nucleus) | Variable based on cell type and context [1] |
Parameter-Specific Methodologies and Experimental Protocols
Orientational Order Parameter (OOP) and COOP
Theoretical Foundation and Calculation: The Orientational Order Parameter (OOP) is derived from liquid crystal physics and provides a quantitative measure of the degree of alignment within a system of pseudo-vectors representing biological structures such as cytoskeletal fibers [29] [30]. The parameter is calculated through tensor analysis of vector orientation data. For a set of pseudo-vectors K, the order tensor is defined as:
𝕋K = 2⟨ki,xki,x⟩⟨ki,xki,y⟩⟨ki,xki,y⟩⟨ki,yki,y⟩ - 𝕀
where 𝕀 is the identity matrix, and the OOP is the maximum eigenvalue of this tensor [30]. The Co-Orientational Order Parameter (COOP) extends this framework to measure correlation between two different constructs (P and Q) by creating a new field F representing the angle between corresponding vectors:
COOPPQ = ⟨fi,x²⟩ + ⟨fi,y²⟩ - 1 + √(⟨fi,x²⟩ - ⟨fi,y²⟩)² + 4⟨fi,xfi,y⟩²
where fi,x = pi·qi and fi,y = |pi×qi| [30]. This enables quantification of how well the orientation of one biological construct (e.g., actin filaments) correlates with another (e.g., Z-lines).
Experimental Workflow for Cytoskeletal Analysis:
- Sample Preparation and Imaging: Culture cells on appropriate substrates, fix at relevant time points, and perform immunofluorescence staining for cytoskeletal components (e.g., α-tubulin for microtubules). Acquire high-resolution Z-stack images using confocal microscopy [1].
- Image Preprocessing: Apply deconvolution to remove noise and blur, followed by maximum intensity projection (MIP) to create 2D images. Process images with Gaussian filtering to smooth fluorescence signal, Sato filtering to highlight curvilinear structures, and Hessian filtering to generate binary images [1].
- Fiber Skeletonization: Convert binary images to skeletonized representations using thinning algorithms to reduce fibers to single-pixel width representations while preserving topology [1].
- Orientation Extraction: Calculate local orientation vectors using line segment detection or gradient-based methods. For each detected fiber segment, compute the orientation angle relative to a reference axis.
- OOP/COOP Computation: Implement the tensor-based calculations described above using computational platforms such as MATLAB or Python. The COOP is particularly valuable for assessing coordination between different cytoskeletal elements in engineered cardiac tissues, where perfect correlation between actin filaments and Z-lines is expected [29].
Figure 1: Experimental workflow for Orientational Order Parameter analysis
Fractal Dimension and Connectivity Metrics
Theoretical Foundation of Fractal Analysis: Fractal dimension quantifies the structural complexity and space-filling characteristics of biological structures by measuring how detail changes with scale. The Modified Blanket Method (MBM) operates by interpreting images as topographical maps and calculating the local surface area as a function of image resolution [31]. The power law relationship between surface area (SA) and pixel size defines the fractal dimension:
FD = 2 - Δlog(SA)/Δlog(pixel size)
This approach enables computation of local FD values at each pixel, facilitating the analysis of specific structures or individual cells [31]. For connectivity assessment, the Local Connected Fractal Dimension (LCFD) provides a specialized metric for evaluating interconnection density within cellular structures.
Experimental Protocol for Fractal Analysis:
- Image Acquisition: Obtain high-quality images of biological structures of interest (e.g., mitochondrial networks, cancer cell nuclei, or cytoskeletal patterns) using appropriate microscopy techniques. For mitochondrial analysis, NADH autofluorescence images can be utilized [31].
- Region of Interest Definition: Delineate precise regions of interest (ROIs) for analysis. For intracellular structures, accurate cell segmentation is crucial, potentially employing clone stamping to fill backgrounds with intracellular feature copies [31].
- MBM Implementation:
- Resample both the image and convolution kernel proportionally
- Compute horizontal and vertical gradients using X- and Y-gradient kernels ([1,-1,0] and [0,-1,1]ᵀ respectively)
- Sum absolute gradient values by convolving with a binary disk kernel
- Calculate local surface area maps by adding horizontal and vertical gradients
- Resample SA maps back to original dimensions
- Repeat with progressively reduced kernel sizes (decrementing by 1 pixel until 1×1 kernel remains)
- Determine FD from the power law exponent relating local SA measurements to pixel sizes [31]
- Validation and Calibration: Assess algorithm accuracy using simulated cell images with known fractal dimension. Validate against established methods like Power Spectral Density (PSD) analysis, which computes a radial average of the 2D Fourier transform of the image [31].
Application to Cancer Cell Nuclei: In prostate cancer diagnostics, fractal analysis of cancer cell nuclei spatial distribution has demonstrated significant diagnostic value. Specific cut-off values for global fractal capacity dimension (D0) enable stratification of carcinomas into distinct aggressiveness classes [32]:
- D0 < 1.5820: Low complexity, low aggressive carcinomas
- D0 > 1.6980: High complexity, high aggressive carcinomas
Complementary parameters including LCFD, lacunarity (λ), and Shannon entropy (H) further refine this classification, with high-aggression carcinomas typically showing LFD > 1.7644, LCFD > 1.7051, H > 0.9, and λ < 0.7 [32].
Figure 2: Fractal dimension analysis workflow using the Modified Blanket Method
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Computational Tools for Organizational Analysis
| Category | Specific Reagents/Tools | Application Purpose | Key Features |
|---|---|---|---|
| Imaging Reagents | α-tubulin antibodies [1] | Microtubule visualization | Enables cytoskeletal architecture analysis |
| Phalloidin conjugates | Actin filament staining | Highlights actin cytoskeleton organization | |
| Hoechst 33342 [33] | Nuclear staining | Defines nuclear boundaries for spatial reference | |
| Computational Tools | MATLAB with customized algorithms [31] [1] | Implementation of MBM, OOP/COOP calculations | Flexible programming environment for custom analysis |
| CellProfiler [33] | Image analysis and segmentation | Open-source platform for high-throughput screening | |
| Topcat [33] | Data visualization and plotting | Specialized tools for biological data representation | |
| Reference Materials | Simulated cell images with known FD [31] | Algorithm validation | Provides ground truth for methodological verification |
| Population-based input functions [34] | Calibration and normalization | Enables cross-study comparisons |
Integration of Multiple Parameters for Enhanced Classification
The most powerful applications of these quantitative parameters emerge when they are integrated to create multidimensional organizational profiles. Research demonstrates that combining fractal dimension with complementary metrics like lacunarity and entropy significantly enhances the discrimination between aggressive and non-aggressive cancer phenotypes [32]. Similarly, the concurrent analysis of orientational order with morphometric parameters (e.g., fiber length, compactness, and radiality) provides a more comprehensive understanding of cytoskeletal reorganization in invasive cells [1].
This integrated approach is particularly valuable in the context of cytoskeletal organization in invasive versus non-invasive cells. Studies have revealed that cancer cells with disrupted E-cadherin and increased invasive potential display distinctive cytoskeletal architecture characterized by shorter microtubules, dispersed orientations (lower OOP), and more compact distribution compared to their non-invasive counterparts [1]. These organizational differences, quantifiable through the parameters described in this guide, provide objective markers of invasive potential that could complement traditional histopathological analysis.
The implementation of these quantitative parameters continues to evolve with advancements in computational power and imaging technology. Future directions include the development of standardized reference datasets, improved algorithms for three-dimensional organizational analysis, and integration with machine learning approaches for automated classification of cellular phenotypes based on organizational signatures.
In vitro cell culture models represent a foundational technology in cancer research, particularly for preclinical drug development. However, traditional two-dimensional (2D) monolayer cultures grown on rigid plastic substrates fail to replicate the complex and dynamic nature of the tumor microenvironment (TME). This microenvironment is a sophisticated ecosystem comprising cancer cells, stromal cells, immune components, endothelial cells, signaling molecules, and the extracellular matrix (ECM). The limitations of 2D models contribute to a significant translational gap, with approximately only 10% of anticancer drug candidates that show promise in conventional cultures progressing successfully to clinical trials [35].
The transition to three-dimensional (3D) culture models addresses these deficiencies by providing a platform that more accurately mimics the structural, biochemical, and cellular interactions found in vivo. These advanced models, including multicellular spheroids, organoids, and bioprinted constructs, recapitulate critical tumor characteristics such as oxygen and nutrient gradients, cell-ECM interactions, and the development of drug-resistant phenotypes [36] [37] [35]. This guide provides a comparative analysis of leading 3D culture technologies, supported by experimental data and detailed methodologies, to inform their application in cancer research and drug development.
Comparative Analysis of 3D Culture Technologies
Various 3D culture technologies have been developed, each with distinct advantages, limitations, and suitability for specific research applications. The table below provides a structured comparison of the primary scaffold-free and scaffold-based methods.
Table 1: Comparison of Primary 3D Cell Culture Technologies
| Technology | Classification | Key Features | Throughput | Physiological Relevance | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|---|
| Hanging Drop [35] | Scaffold-free | Cells aggregate at liquid-air interface to form spheroids | Medium | Medium - Recapitulates cell-cell contacts; lacks ECM | Produces uniform, tightly-packed spheroids; no artificial scaffold | Difficult medium changes; limited spheroid size; absent cell-ECM interactions |
| Forced Floating [35] | Scaffold-free | Cells form spheroids on ultra-low attachment surfaces | High | Medium - Recapitulates cell-cell contacts; lacks ECM | Simple protocol; compatible with various assays; uniform spheroid size | Absent cell-ECM interactions; does not fully mimic tumor physiology |
| Bioreactors [35] | Scaffold-free | Spheroid formation via agitated suspension culture | High (for large quantities) | Medium - Recapitulates cell-cell contacts; lacks ECM | Generates large spheroid quantities; long-term viability | Significant spheroid size variation; potential shear stress damage |
| Organoids [36] [38] | Scaffold-based | Stem cells differentiate into 3D, organ-like structures in a matrix (e.g., Matrigel) | Low to Medium | High - Retains tumor heterogeneity; contains multiple cell types | Preserves patient-specific genetics and tumor heterogeneity; high clinical predictive value | Technically challenging; culture instability; batch-to-batch matrix variability |
| 3D Bioprinting [39] | Scaffold-based | Layer-by-layer deposition of bioinks (cells + biomaterials) to create complex structures | Medium (Rapidly improving) | High - Customizable architecture; can incorporate vasculature and multiple cell types | High precision and control over TME composition and spatial organization; rising integration with AI for optimization | High cost; requires specialized equipment; complex protocol optimization (bioink, parameters) |
| Tumor-on-Chip [37] | Scaffold-based or free | Microfluidic devices culture cells in 3D gels or as spheroids under perfused flow | Low to Medium | High - Mimics fluid flow, shear stresses, and systemic delivery | Enables real-time monitoring of metabolites (e.g., glucose, lactate); models delivery dynamics | Specialized device design and operation; lower throughput |
Quantitative Data from 2D vs. 3D Model Comparisons
Empirical studies consistently demonstrate significant phenotypic and functional differences between cells cultured in 2D versus 3D formats. The following tables summarize key experimental findings that highlight the superior biological relevance of 3D models.
Table 2: Phenotypic and Behavioral Differences Between 2D and 3D Cultures
| Parameter | 2D Culture | 3D Culture | Research Implications |
|---|---|---|---|
| Cell Morphology [40] | Flat, stretched | In vivo-like, often spherical | 3D morphology influences cytoskeletal organization, cell polarity, and mechanotransduction. |
| Proliferation Rate [36] [37] | Rapid, contact-inhibited | Slower, more in vivo-like | Models tumor dormancy and quiescent cell populations often resistant to therapy. |
| Gene Expression [40] [37] | Altered profiles; functional simplification | Closer to in vivo profiles; unique regulatory patterns | Drug targets and signaling pathway activity in 2D may not reflect the in vivo tumor. |
| Cell Communication [36] | Primarily cell-cell | Cell-cell, cell-matrix, and spatial interactions | Critical for studying invasion, metastasis, and resistance mechanisms. |
| Drug Response [37] [35] | Often more sensitive | Increased resistance due to gradients and ECM protection | More accurately predicts in vivo drug efficacy and penetration challenges. |
| Metabolic Patterns [37] | Uniform nutrient access | Gradients leading to heterogeneous metabolic zones | Models regions of hypoxia and metabolic adaptation within tumors. |
Table 3: Experimental Data from Direct Comparisons of 2D and 3D Models
| Study Focus | Cell Line/Model | Key Finding in 3D vs. 2D | Quantitative Result | Citation |
|---|---|---|---|---|
| Proliferation & Metabolism | U251-MG (Glioblastoma) & A549 (Lung) | Reduced proliferation in 3D, especially under glucose restriction. Higher per-cell glucose consumption. | In 3D, cells survived >5 days without glucose; in 2D, death occurred by day 3. 3D models showed elevated lactate production. | [37] |
| Transcriptomics | A549 (Cancer) & BEAS-2B (Normal) | Significant differential gene expression related to cell cycle, immune response, and cell adhesion. | Key genes ACTB, FN1, and IL6 identified as potential hubs in 3D organoid formation. | [40] |
| Drug Sensitivity | OVCAR3-based Tumoroids | Tumoroids with non-tumor stromal/immune cells showed higher viability post-treatment than cancer-only spheroids. | Cancer-only spheroids (Composition 1) showed lowest viability (0.563) after carboplatin vs. spheroids with U937 monocytes (0.954). | [41] |
| Radiation Response | A549 Organoids | 3D-grown cells demonstrated increased radio-resistance compared to 2D cultured cells. | - | [40] |
Experimental Protocols for Key 3D Models
Protocol 1: Establishing a High-Throughput Tumoroid Co-Culture Model for Drug Screening
This protocol, adapted from a study predicting TME-mediated chemoresistance, details the creation of tumoroids with defined cellular compositions to investigate the impact of stromal and immune cells on drug response [41].
Step 1: Cell Preparation
- Harvest and count all cell types. The model uses OVCAR3 (high-grade serous ovarian cancer cells), Mesenchymal Stem Cells (MSCs), HUVECs (human umbilical vein endothelial cells), and U937 monocytes.
- Create a master cell suspension mix for each of the 23 desired compositions as defined in the experimental design (e.g., Composition 1: 1000 OVCAR3; Composition 11: 1000 OVCAR3 + 500 U937).
Step 2: Tumoroid Formation via Forced Floating
- Transfer cell suspension aliquots (e.g., 100 µL containing the total cell number for one spheroid) to the wells of a 96-well ultra-low attachment (ULA) plate.
- Centrifuge the plate at a low speed (e.g., 300-500 x g for 5 minutes) to promote cell aggregation at the bottom of the well.
- Incubate the plate at 37°C with 5% CO₂ for 48-72 hours to allow for spheroid formation and stabilization.
Step 3: Drug Treatment and Viability Assay
- After spheroid formation, add chemotherapeutics (e.g., carboplatin, paclitaxel) or targeted agents to the wells at the desired concentrations.
- Incubate for 48 hours.
- Assess cancer cell viability using a standard MTS assay. Add the MTS reagent to each well, incubate for several hours, and measure the absorbance at 490 nm. Normalize viability values to untreated control tumoroids of the same composition.
Step 4: Data Analysis and Machine Learning Integration
- Analyze the viability data to correlate tumoroid cellular composition with drug response.
- For predictive modeling, input the composition data (number of each cell type per spheroid) and the corresponding viability outcomes into a Random Forest machine learning algorithm to identify which non-tumor cell types most significantly drive chemoresistance [41].
Protocol 2: Generating 3D Organoids from Patient-Derived Cells
Patient-derived tumor organoids (PDTOs) preserve the genetic and phenotypic heterogeneity of the original tumor, making them powerful tools for personalized medicine [36] [38].
Step 1: Tissue Digestion
- Obtain fresh patient tumor tissue via biopsy or surgery, under informed consent and ethical approval.
- Mechanically mince the tissue into small fragments (~0.5-1 mm³) and enzymatically digest using a cocktail of collagenase and dispase for 30-60 minutes at 37°C.
Step 2: Cell Extraction and Seeding
- Filter the digested suspension through a cell strainer (70-100 µm) to remove undigested fragments and obtain a single-cell suspension.
- Centrifuge to pellet the cells and resuspend in a cold, growth factor-reduced Basement Membrane Extract (BME, e.g., Matrigel). The BME mimics the native extracellular matrix.
Step 3: 3D Culture and Maintenance
- Plate small droplets (e.g., 20-50 µL) of the cell-BME suspension into pre-warmed tissue culture plates. Invert the plate to create a hanging drop setup or simply pipette onto the surface.
- Culture the plates at 37°C for 20-30 minutes to allow the BME to polymerize into a solid gel.
- Carefully overlay the polymerized droplets with organoid-specific culture medium. This medium is typically supplemented with a tailored mix of growth factors (e.g., Wnt3A, R-spondin, Noggin, EGF) to support stem cell expansion and inhibit fibroblast overgrowth [38].
- Refresh the medium every 2-3 days. Organoids can be passaged every 1-2 weeks by mechanically and enzymatically breaking them into smaller fragments and re-embedding them in fresh BME.
Step 4: Organoid-Immune Co-Culture
- To model immune interactions, establish a co-culture system. Isate tumor-infiltrating lymphocytes (TILs) from the same patient's tissue.
- Once organoids are established, add the isolated TILs directly to the culture well. This model can be used to study immune checkpoint blockade (e.g., anti-PD-1/PD-L1) and other immunotherapies ex vivo [38].
Signaling Pathways and Molecular Mechanisms in 3D Microenvironments
The 3D architecture profoundly influences cellular signaling. Transcriptomic analyses reveal that cells in 3D cultures exhibit unique gene regulatory patterns compared to their 2D counterparts, activating pathways related to cell adhesion, immune response, and matrix interaction [40]. The following diagram illustrates key signaling pathways and cellular interactions that are more accurately recapitulated in 3D TME models.
Diagram 1: Key signaling pathways and cell-cell interactions in the 3D tumor microenvironment. Pathways in yellow represent key molecular players (e.g., IL-6, ACTB) upregulated in 3D cultures. Blue nodes represent non-tumor cells in the TME that drive resistance. Red nodes represent tumor cell outcomes. Green nodes represent ECM-mediated signaling. CSC: Cancer Stem Cell.
The Scientist's Toolkit: Essential Reagents and Solutions
Successful implementation of advanced 3D models requires specific reagents and materials. The following table details key solutions used in the protocols and studies cited in this guide.
Table 4: Essential Research Reagent Solutions for 3D Culture Models
| Reagent/Material | Function/Application | Specific Examples & Notes |
|---|---|---|
| Basement Membrane Extracts (BME) | Provides a biologically active scaffold that mimics the in vivo extracellular matrix for scaffold-based cultures. | Matrigel (Corning), Cultrex BME (R&D Systems). Note: Batch-to-batch variability is a known challenge. |
| Synthetic Hydrogels | Defined, reproducible synthetic ECM; allows precise control over mechanical and biochemical properties. | Gelatin Methacrylate (GelMA), Polyethylene Glycol (PEG)-based hydrogels. |
| Ultra-Low Attachment (ULA) Plates | Prevents cell adhesion to the plastic surface, forcing cells to aggregate and form spheroids in scaffold-free methods. | Corning Spheroid Microplates, Nunclon Sphera plates. |
| Specialized Growth Factors | Promotes the growth and maintenance of specific cell types (especially stem/progenitor cells) in organoid cultures. | Wnt3a, R-spondin, Noggin, EGF, FGF10, B27 supplement. The cocktail is tissue-specific [38]. |
| Cell Viability Assays | Measures metabolic activity or ATP content as a proxy for cell viability and proliferation in 3D structures. | MTS, CCK-8, CellTiter-Glo 3D. Note: Standard assays may require optimization for 3D cultures. |
| Microfluidic Devices | Provides a platform for "Tumor-on-Chip" models, enabling perfusion, mechanical stress, and real-time metabolite monitoring. | Custom designs or commercial systems (e.g., from Emulate, AIM Biotech). |
| Bioinks | Formulations of living cells and biomaterials used as "inks" in 3D bioprinting to create tissue constructs. | Typically blends of hydrogels (e.g., alginate, GelMA, hyaluronic acid) and cells [39]. |
Emerging Trends and Future Perspectives: The Role of AI and Advanced Imaging
The field of 3D cancer modeling is rapidly evolving, with several cutting-edge technologies enhancing its predictive power.
Integration with Artificial Intelligence (AI): AI and machine learning are being leveraged to optimize 3D bioprinting parameters, analyze complex high-dimensional data from 3D models, and predict drug responses. For instance, random forest algorithms can predict TME-driven chemoresistance based on tumoroid cellular composition [41]. Furthermore, the combination of mechanistic computational modeling (e.g., agent-based models of the TME) with AI is paving the way for creating patient-specific "digital twins" to simulate treatment outcomes and optimize therapy selection [42] [43].
Multi-omics and High-Throughput Screening: The combination of 3D models with high-throughput sequencing (e.g., RNA-seq) allows for the discovery of 3D-specific gene expression signatures, as demonstrated in A549 and BEAS-2B cells [40]. High-content imaging and automated analysis platforms are also being developed to scale up 3D drug screening.
Advanced Imaging and Metabolic Monitoring: Technologies like Seahorse XF Analyzers and D-OCT are being incorporated to monitor metabolic flux and spatial organization in real-time within 3D cultures [36]. Tumor-on-chip platforms enable continuous, non-invasive monitoring of metabolite consumption (glucose, glutamine) and waste production (lactate), providing dynamic insights into tumor metabolism [37].
In conclusion, advanced 3D culture models represent a paradigm shift in cancer research, offering a more physiologically relevant context for studying tumor biology, stromal interactions, and drug efficacy. While challenges in standardization and scalability remain, the integration of these models with AI, multi-omics, and advanced engineering promises to significantly accelerate the development of more effective, personalized cancer therapies.
Quantitative Phase Imaging (QPI) represents a paradigm shift in live-cell analysis, enabling non-invasive, label-free quantification of cellular kinetics and morphology. This technology is particularly transformative for research into cytoskeletal organization and its role in cellular invasion, as it allows continuous monitoring of dynamic processes without fluorescent labels that can cause phototoxicity or alter native cell behavior. By measuring phase shifts in light passing through cells, QPI generates quantitative data on cell dry mass, morphology, and dynamic movements, providing a powerful tool for comparing invasive and non-invasive cell phenotypes. The integration of QPI with machine learning has further enhanced its capability to predict functional cell outcomes based on temporal kinetic data, advancing from static snapshots to dynamic forecasting of cellular behavior. This guide objectively compares QPI's performance against other label-free methodologies within the specific context of cytoskeletal research.
Experimental Protocols for Cytoskeletal and Invasion Studies
QPI Protocol for Single-Cell Kinetic Analysis of Stem Cells
This protocol, adapted from hematopoietic stem cell (HSC) research, details how QPI captures cytoskeleton-driven cellular dynamics [44].
Cell Culture and Preparation: Sort single CD201+CD150+CD48−KSL murine hematopoietic stem cells or Lin-CD34+CD38-CD45RA-CD90+CD201+ human HSCs via fluorescence-activated cell sorting (FACS). Culture individual cells in 96-well U-bottom plates using established ex vivo expansion media that maintains stemness. Maintain cells in a stage-top incubator ensuring stable conditions (37°C, 5% CO₂) throughout imaging.
Image Acquisition: Use a holographic imaging system (e.g., HoloMonitor or Phasics SID4-Bio camera) integrated with an inverted microscope. Employ a 20× objective (NA 0.4) for a balance between resolution and field of view. Acquire time-lapse images every 5-15 minutes for 72-96 hours using white light illumination to ensure minimal phototoxicity. The system captures interferograms, which are computationally reconstructed into quantitative phase images.
Data Extraction and Analysis: Process phase images to segment and track individual cells across all time points. Extract kinetic parameters including:
- Cellular Dry Mass: Calculated from the integrated phase shift across the cell area.
- Morphological Parameters: Cell area, contour length, and sphericity.
- Motility Metrics: Displacement velocity and directorial persistence.
- Proliferation Kinetics: Division timing, division symmetry, and cytokineis patterns.
- Advanced Analysis: Utilize Uniform Manifold Approximation and Projection (UMAP) on extracted features to identify distinct cell clusters based solely on kinetic behavior.
Computational Pipeline for Cytoskeletal Architecture Analysis
This protocol details a bioimage informatics approach for quantifying cytoskeletal organization from fluorescence images, providing a complementary method to QPI [1].
Sample Preparation and Imaging: Culture cells (e.g., wild-type vs E-cadherin mutant invasive variants) on laminin-coated coverslips. Fix, permeabilize, and immunostain for α-tubulin (microtubules) and nucleus (DAPI). Acquire high-resolution Z-stack images using a fluorescence microscope with a 63× or 100× oil-immersion objective. Deconvolve images to reduce noise and improve resolution.
Image Processing and Feature Extraction: Process deconvolved maximum intensity projections through a Gaussian filter for smoothing, followed by a Sato filter to highlight curvilinear structures of cytoskeletal fibers. Apply Hessian-based filtering and thresholding to generate binary images of the cytoskeletal network. Skeletonize binary images to create a 1-pixel-wide representation of each fiber.
Quantitative Cytoskeletal Metrics: Extract two classes of features from the skeletonized images:
- Line Segment Features (LSFs): Quantify fiber orientation (Orientational Order Parameter), length, and quantity.
- Cytoskeleton Network Features (CNFs): Assess topology through compactness (fibers/area), radiality (radial score relative to nucleus centroid), bundling, parallelism, connectivity, and complexity (fractal dimension).
Electrical Impedance Spectroscopy (EIS) for Cell Dynamics Monitoring
This protocol describes EIS as an alternative, non-microscopic method for monitoring cell behavior [45].
Platform Setup: Use a microfabricated microelectrode array (MEA) platform with 25 electrode pairs. Culture cells directly on the electrode surface. For coculture invasion models, use bilateral or concentric seeding configurations with normal and cancerous cell types.
Impedance Measurement and Correlation: Acquire EIS spectra continuously across a frequency range (e.g., 10 Hz - 2 MHz) from all electrode positions. Simultaneously, acquire bright-field and fluorescence microscopy images adjacent to each electrode pair at regular intervals. Correlate impedance measurements (modulus and phase) with cellular parameters extracted from image segmentation.
Machine Learning Integration: Train a deep learning model using paired EIS and image segmentation data. Use the model to predict spatiotemporal evolution of cell density, covered area, mean cell diameter, and cell type classification based solely on EIS measurements.
Technology Performance Comparison
The table below summarizes the quantitative performance of QPI against other label-free technologies in the context of cytoskeletal and invasion research.
Table 1: Performance Comparison of Label-Free Live-Cell Monitoring Technologies
| Technology | Spatial Resolution | Temporal Resolution | Key Measurable Parameters | Cytoskeletal Insight | Throughput |
|---|---|---|---|---|---|
| Quantitative Phase Imaging (QPI) | Sub-micrometer (diffraction-limited) [46] | Seconds to minutes (10 fps demonstrated) [47] | Dry mass, morphology, motility, proliferation kinetics [44] [47] | Indirect via cell morphology and mass distribution; reveals division anomalies [44] | Medium (multi-well imaging) [47] |
| Brightfield Microscopy | Sub-micrometer (diffraction-limited) [46] | Seconds to minutes [46] | Confluency, basic morphology, migration [46] | Limited to gross morphological changes | High (standard equipment) [46] |
| Electrical Impedance Spectroscopy (EIS) | Millimeter to centimeter (electrode-dependent) [45] | Minutes to hours [45] | Cell density, covered area, cell-cell interactions [45] | No direct structural information; infers cytoskeletal changes through behavior | High (real-time, multiplexed electrodes) [45] |
| Microfluidic Broadband EIS | Single-cell (with trapping) [48] | Minutes (per single cell) [48] | Cytoplasm conductivity, membrane integrity, viability [48] | No direct structural information | Low (sequential single-cell analysis) [48] |
Table 2: Quantitative Cytoskeletal and Kinetic Parameters from Featured Studies
| Parameter Category | Specific Metric | Invasive/ Mutant E-cadherin Cells | Non-Invasive/ Wild-Type Cells | Measurement Technology |
|---|---|---|---|---|
| Microtubule Architecture | Orientational Order Parameter (OOP) | Significantly lower (disorganized fibers) [1] | Higher (well-aligned fibers) [1] | Fluorescence microscopy + computational analysis [1] |
| Microtubule Architecture | Fiber Compactness (Nl/Ac) | More compactly distributed [1] | More dispersed distribution [1] | Fluorescence microscopy + computational analysis [1] |
| Microtubule Architecture | Fiber Length (LiE) | Shorter fibers [1] | Longer fibers [1] | Fluorescence microscopy + computational analysis [1] |
| Cellular Kinetics | Proliferation Rate | Heterogeneous, some with rapid initial proliferation then differentiation [44] | Slower initial proliferation but sustained over time [44] | QPI [44] |
| Cellular Kinetics | Division Pattern | 8.21% showed cytokinesis interruption [44] | 91.3% normal division patterns [44] | QPI [44] |
| Cell Morphology | Dry Mass | Heterogeneous output (10.9% produced cells >200 pg) [44] | More consistent dry mass distribution [44] | QPI [44] |
Signaling Pathways and Experimental Workflows
Diagram 1: Experimental workflow for label-free comparison of invasive and non-invasive cells. The pathway integrates QPI, EIS, and cytoskeletal analysis with machine learning to classify cell phenotypes based on kinetic and structural features.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Reagent Solutions for Label-Free Live-Cell Assays
| Reagent/Solution | Function/Purpose | Example Application |
|---|---|---|
| Low-Conductivity Sucrose/Dextrose Isotonic Solution | Provides physiological osmolarity while minimizing electrical interference in EIS [48] | EIS measurements of single yeast cells; maintains viability with minimal signal distortion [48] |
| Lectin Coating | Enhances cell adhesion to electrode surfaces in microfluidic EIS devices [48] | Improves single-cell trapping and measurement stability in CPW-based EIS platforms [48] |
| Specialized HSC Expansion Media | Supports ex vivo stem cell proliferation while maintaining stemness potential [44] | Long-term kinetic tracking of hematopoietic stem cell divisions and fate decisions using QPI [44] |
| Laminin-Coated Substrata | Mimics extracellular matrix for studies of cell-ECM interactions in invasion models [1] | Provides physiological context for cytoskeletal architecture analysis in invasive vs non-invasive cells [1] |
| Coculture Media Mixtures | Supports multiple cell types in coculture invasion models [45] | Enables EIS monitoring of cell competition and spatial heterogeneity in normal/cancer cell cocultures [45] |
Quantitative Phase Imaging establishes a powerful standard for non-invasive kinetic analysis in cytoskeletal and invasion research, providing unparalleled single-cell resolution and rich temporal data on cellular dynamics. While EIS offers superior temporal monitoring for population-level behaviors and electrical properties, it lacks direct structural insight into cytoskeletal organization. The complementary application of these technologies, enhanced by computational analysis and machine learning, creates a robust framework for distinguishing invasive from non-invasive cellular phenotypes based on their distinct kinetic and structural signatures. This multi-modal approach advances our capacity to predict cellular behavior and functional outcomes from dynamic, label-free observations.
Impedance-Based Real-Time Analysis (e.g., xCELLigence) for Dynamic Motility Profiling
The study of cell motility, a process fundamental to cancer metastasis, immune response, and tissue repair, has traditionally relied on endpoint assays that provide static snapshots of dynamic processes. Impedance-based real-time cell analysis (RTCA), commercially available as the xCELLigence system, represents a transformative technological approach that enables continuous, label-free monitoring of cellular behaviors including proliferation, migration, and invasion [49] [50]. This guide provides a comprehensive comparison of xCELLigence performance against conventional methodologies, with particular emphasis on its application in cytoskeletal research for distinguishing invasive and non-invasive cellular phenotypes.
The xCELLigence system operates on the principle of measuring electrical impedance across microelectrodes integrated into the bottom of culture plates [49]. As cells adhere, spread, and proliferate on these electrodes, they impede the flow of electrical current in proportion to their biological status. This impedance is automatically converted into a unitless parameter called the Cell Index (CI), which quantitatively reflects cell number, size, shape, and adhesion quality [49] [51]. The system's ability to monitor these parameters without labels or invasive procedures makes it particularly valuable for long-term kinetic studies of cellular processes that involve cytoskeletal rearrangements [49].
Technology Comparison: xCELLigence Versus Conventional Methods
Performance Benchmarking Against Established Techniques
Table 1: Comparative Analysis of xCELLigence and Conventional Cell Analysis Methods
| Analysis Parameter | Conventional Method | Correlation with xCELLigence | Key Advantages of xCELLigence |
|---|---|---|---|
| Cell Proliferation | Sulforhodamine B (SRB) assay | Spearman's ρ = 0.688 - 0.964 [50] | Real-time kinetics, label-free, non-destructive |
| Cytotoxicity | SRB assay | Spearman's ρ > 0.95 [50] | Continuous monitoring, identifies transient responses |
| Cell Migration | Transwell/Boyden assay | Spearman's ρ = 0.90 (area calculation), ρ > 0.95 (OD reading) [50] | Kinetic data, no membrane transfer required |
| Cell Invasion | Matrigel-coated Transwell | Spearman's ρ > 0.95 [50] | Real-time monitoring of complex invasion process |
| Barrier Function | Trans-epithelial electrical resistance (TEER) | Non-invasive, automated measurement without electrode positioning variability [49] | Continuous assessment, no operator-dependent variability |
When compared to traditional endpoint assays, xCELLigence demonstrates strong statistical correlation while providing superior temporal resolution. For proliferation assessment, studies with A549 and MDA-MB-231 cell lines revealed correlation coefficients with SRB assays ranging from 0.688 to 0.964 depending on cell seeding density [50]. Similarly, cytotoxicity testing with paclitaxel showed nearly identical IC50 values between xCELLigence and SRB methods (4.78±0.90 nM vs. 6.44±1.90 nM respectively) with correlation coefficients exceeding 0.95 [50].
For motility studies, the conventional Transwell migration assay requires laborious manual counting or staining procedures that capture only a single time point. In contrast, xCELLigence provides continuous kinetic data throughout the experiment, revealing migration patterns that might be missed with endpoint approaches [50]. The technology's particular strength lies in its ability to monitor barrier function in epithelial models like Caco-2 cells, overcoming critical limitations of traditional TEER measurements which require repeated electrode insertion and are susceptible to operator-dependent variability [49].
Practical Implementation Considerations
Table 2: Methodological Requirements and Practical Considerations
| Parameter | xCELLigence System | Conventional Methods |
|---|---|---|
| Throughput | Medium to high (16, 96, or 384-well formats available) [49] | Variable (typically lower for kinetic studies) |
| Assay Duration | Continuous (hours to days) | Endpoint (multiple discrete time points required) |
| Cell Type Adaptability | Requires optimization for non-adherent cells (e.g., fibronectin coating) [51] | Broad applicability with protocol modifications |
| Data Output | Real-time Cell Index (CI) values | Variable (absorbance, fluorescence, cell counts) |
| Labor Intensity | Low after initial setup | High (multiple manual steps) |
| Cost Considerations | Higher initial instrument investment | Lower initial cost, higher recurrent reagent costs |
While xCELLigence offers significant advantages for temporal monitoring, its implementation requires consideration of several factors. The system requires adherent cell types or conditions that promote adhesion, as impedance detection depends on cell-electrode contact [51]. For non-adherent hematopoietic cells, pre-coating E-plates with extracellular matrix components like fibronectin has proven effective, significantly increasing cell attachment compared to collagen, gelatin, or laminin substrates [51]. The initial financial investment for xCELLigence instrumentation is substantial, though this may be offset by reduced reagent costs and labor savings over time.
Experimental Design for Motility Profiling
Protocol: Real-Time Monitoring of 2D Cell Motility and Cytoskeletal Function
The following protocol outlines the optimized methodology for implementing xCELLigence technology in motility and cytoskeletal studies:
Instrument Setup: Initialize the xCELLigence RTCA station placed inside a standard tissue culture incubator (37°C, 5% CO2). Install the appropriate E-plate (16, 96, or 384-well format) and establish communication with the control software [49] [51].
Background Measurement: Add 50-100 μL of cell culture medium to each well and perform initial impedance scanning to establish baseline values. This critical step ensures proper electrode function and identifies any well-to-well variability [51] [52].
Cell Seeding and Adhesion Monitoring: Seed cells at optimized densities (typically 5,000-40,000 cells/cm² depending on cell type) in a total volume of 100-200 μL. Continuously monitor impedance every 1-15 minutes during the initial adhesion phase (4-24 hours) to establish quality of cell attachment [51] [52].
Experimental Intervention: Once cells reach desired confluence (typically CI plateau), add compounds targeting cytoskeletal elements (e.g., paclitaxel, vinorelbine) or migration modulators. Include appropriate vehicle controls in parallel wells [50] [53].
Real-Time Data Acquisition: Monitor Cell Index every 15-60 minutes for duration of experiment (typically 24-72 hours). The frequency can be adjusted based on the anticipated kinetics of the biological response [50].
Data Analysis: Normalize Cell Index values to pre-treatment time points or control wells. Analyze kinetic parameters including time to peak CI, CI slope, and area under the curve [50].
For non-adherent cell types (e.g., leukemia/lymphoma cells), include a pre-coating step with fibronectin (6 μg/well for 1 hour at 37°C) followed by washing with PBS prior to cell seeding [51].
Protocol: 3D Migration and Invasion Assessment
The CIM-Plate (Cell Invasion and Migration Plate) system enables real-time monitoring of invasion through extracellular matrix barriers:
Matrix Coating: Dilute Matrigel or other ECM substrates in serum-free medium and add to the upper chamber of the CIM-Plate. Allow to gel at 37°C for 1-4 hours [50].
Cell Preparation: Harvest and resuspend cells in serum-free medium. Seed 30,000-100,000 cells in the upper chamber depending on cell type and invasion capacity.
Chemoattractant Application: Add medium containing chemoattractant (e.g., 10% FBS) to the lower chamber. Serum-free medium in both chambers serves as a negative control for random migration [50].
Assembly and Monitoring: Assemble the CIM-Plate and place in the xCELLigence station. Monitor impedance every 5-15 minutes as cells migrate through the microporous membrane (8μm pores) toward the chemoattractant gradient [50].
Data Interpretation: The Cell Index directly correlates with the number of cells that have migrated through the membrane and contacted the electrodes on the underside. Compare CI values between Matrigel-coated (invasion) and uncoated (migration) wells to distinguish invasive potential [50].
Figure 1: Experimental workflow for real-time invasion monitoring using CIM-Plates
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Impedance-Based Motility Assays
| Reagent | Function | Application Notes |
|---|---|---|
| E-Plates | Culture plates with integrated gold microelectrodes | Available in 16, 96, and 384-well formats; single-use [49] |
| CIM-Plates | Specialized plates for migration/invasion studies | Feature microporous (8µm) membrane separating upper and lower chambers [50] |
| Fibronectin | Extracellular matrix coating for non-adherent cells | Optimal at 6μg/well; significantly improves adhesion of hematopoietic cells [51] |
| Matrigel | Basement membrane extract for invasion studies | Dilute in serum-free medium; forms 3D barrier in CIM-Plates [50] |
| Paclitaxel | Microtubule-stabilizing cytotoxic agent | Positive control for cytoskeletal disruption; IC50 ~5nM in MDA-MB-231 cells [50] [53] |
| Targeted NPs | Cytoskeleton-disrupting nanoparticles (e.g., ZIF-8) | Inhibit cancer cell migration by altering actin organization [54] |
Cytoskeletal Dynamics and Signaling Pathways
The xCELLigence system provides unique insights into how pharmacological and genetic perturbations affect cytoskeletal organization and consequent motility. The actin cytoskeleton serves as the primary structural framework driving cell motility, with constant F-actin polymerization and depolymerization enabling cells to migrate and invade [54] [4]. Key regulators include the Arp2/3 complex, which drives branched actin polymerization, and formin proteins, which generate linear actin filaments [4]. These elements cooperate to pattern the actin cytoskeleton, with formin-derived actin arcs undergoing myosin II-dependent contraction to transport signaling complexes inward during immune synapse formation or invadopodia function in cancer cells [4].
Metal-organic framework nanoparticles (ZIF-8 NPs) have demonstrated significant effects on cytoskeletal dynamics, altering actin organization through intracellular degradation and subsequent elevation of zinc ion concentrations [54]. When functionalized with EpCAM antibodies, these nanoparticles can specifically target cancer cells and disrupt EpCAM-mediated actin anchoring, significantly inhibiting migration and invasion of breast and prostate cancer cells [54]. The xCELLigence system effectively captures these cytoskeletal alterations through changes in Cell Index, correlating with conventional migration endpoints while providing superior kinetic resolution.
Figure 2: Signaling pathways linking external stimuli to cytoskeletal remodeling
Impedance-based real-time analysis represents a significant advancement over conventional methods for dynamic motility profiling, particularly in the context of cytoskeletal organization research. The xCELLigence system provides unprecedented temporal resolution of cellular processes while maintaining strong correlation with established endpoint assays. Its label-free, non-invasive nature enables long-term kinetic studies that reveal biological responses that would be missed with traditional snapshot approaches.
For researchers investigating the intricate relationship between cytoskeletal dynamics and cellular motility, xCELLigence offers a powerful platform for quantifying how genetic, pharmacological, and microenvironmental factors influence invasive behavior. While the technology requires consideration of cell adhesion properties and represents a substantial initial investment, its ability to provide continuous, quantitative data on motility parameters makes it an invaluable tool for modern cell biology research, particularly in the realms of cancer metastasis, immunology, and drug discovery.
Navigating Technical Challenges: From 2D Artifacts to Cell Line-Specific Variations
The cytoskeleton serves as the mechanical framework of the cell, dynamically orchestrating critical processes including cell division, migration, and invasion. In cancer research, cytoskeletal reorganization is intimately linked with invasive potential and metastatic progression. The choice between two-dimensional (2D) and three-dimensional (3D) culture systems significantly influences the observed architecture and function of this complex scaffold, presenting researchers with a critical methodological conundrum. While 2D monolayers on rigid plastic surfaces have long been the standard for their simplicity and reproducibility, they often generate artifacts that misrepresent native cellular behavior. Conversely, 3D models more accurately mimic the in vivo microenvironment, providing physiologically relevant insights into cytoskeletal organization and its role in cellular invasion. This guide objectively compares these systems, supported by experimental data, to inform best practices in cytoskeletal analysis within cancer research.
Quantitative Comparison of Cytoskeletal Properties in 2D vs. 3D Cultures
Substantial differences in cytoskeletal organization and resulting cellular phenotypes emerge when cells are cultured in 2D versus 3D environments. The table below summarizes key comparative findings from recent studies.
Table 1: Quantitative Differences in Cytoskeletal Organization and Function in 2D vs. 3D Cultures
| Analyzed Parameter | Findings in 2D Culture | Findings in 3D Culture | Reference Model |
|---|---|---|---|
| Actin Fiber Organization | Prominent actin stress fiber bundling [55] | Reduced fiber bundling, relegated to lateral borders of cell extensions [55] | MDA-MB-231 breast cancer cells [55] |
| Traction Forces | Substantially higher traction stresses exerted [56] | Significantly lower traction forces [56] | LN229/T98G glioblastoma cells [56] |
| Metabolic Phenotype | Uniform nutrient access, predominantly proliferative population [37] | Distinct microenvironments; cells at various stages (proliferation, quiescence) [37] | U251-MG glioblastoma & A549 lung adenocarcinoma [37] |
| Invasive Strategy | Not explicitly quantified | Higher amoeboid invasion at distal invasive front with radially oriented matrix [57] | Melanoma and Breast cancer models [57] |
| Microtubule Architecture | More organized, higher Orientational Order Parameter (OOP) [1] | Shorter fibers, disperse orientations, more compact distribution (lower OOP) in invasive cells [1] | E-cadherin mutant invasive cell model [1] |
| Cellular Contractility | Forces correlate with 3D matrix reorganization; disrupted by actin/myosin/MT agents [55] | Force generation mechanisms similar, but magnitude and context differ [55] | MDA-MB-231 breast cancer cells [55] |
Experimental Protocols for Cytoskeletal Analysis
To ensure reproducible and artifact-free analysis, standardized protocols are essential. Below are detailed methodologies for key experiments cited in this guide.
This protocol is used to create a physiologically relevant model for studying cytoskeletal maturation and function.
- Cell Source: Immortalized human skeletal muscle cell line (e.g., AB1167 from MyoLine).
- Scaffold Fabrication:
- 3D print custom polylactic acid (PLA) scaffolds with anchor points (e.g., 5 mm apart).
- Sterilize scaffolds in 70% ethanol and coat overnight at 4°C with 0.2% (w/v) Pluronics F127 to prevent non-specific cell adhesion.
- Embed scaffolds in a thin layer of 4% agarose.
- Hydrogel Cell Encapsulation:
- Mix myoblasts with a hydrogel precursor (e.g., collagen or fibrinogen solution).
- Seed the cell-hydrogel mixture into the scaffold reservoirs, allowing a gel to form around the anchor points.
- Culture and Differentiation:
- Maintain constructs in complete growth medium until confluent.
- Switch to differentiation medium (e.g., DMEM high glucose supplemented with 2% horse serum and 10 µg/mL insulin).
- Change medium every 48 hours for up to 21 days to study maturation.
- Functional Analysis: Passive tension and contractile force can be measured directly from the 3D constructs.
TFM measures the forces cells exert on their substrate, providing insight into cytoskeletal contractility.
- 2D Substrate Preparation:
- Synthesize polyacrylamide (PA) gel substrates with a tunable Young's Modulus (e.g., 5 kPa to mimic physiological stiffness).
- Functionalize gel surfaces with N-6-((acryloyl)amido)hexanoic acid and conjugate with type I collagen (0.1 mg/mL).
- 3D Matrix Preparation:
- Embed cells within a 3D collagen-based matrix (e.g., type I rat tail collagen at 1.3 mg/ml concentration).
- Cell Seeding and Imaging:
- Seed cells sparsely on 2D PA gels or within 3D collagen matrices.
- For 2D TFM, acquire images of fluorescent beads embedded in the gel before and after cell detachment.
- For 3D force assessment, use Confocal Reflectance Microscopy to image and quantify collagen fibril reorganization by single cells.
- Data Analysis:
- Calculate traction stress vectors from the bead displacement field in 2D.
- Model collagen fibril reorganization using an exponential decay model as an indirect measure of single-cell force in 3D.
This image-based pipeline quantifies subtle cytoskeletal alterations associated with invasive potential.
- Sample Preparation and Imaging:
- Culture cells on appropriate 2D or 3D substrates.
- Fix, permeabilize, and stain for cytoskeletal components (e.g., α-tubulin for microtubules, phalloidin for F-actin) and nucleus (DAPI).
- Acquire high-resolution Z-stack images using immunofluorescence and confocal microscopy.
- Image Preprocessing:
- Apply deconvolution to remove noise and blur.
- Create a 2D maximum intensity projection (MIP) of the Z-stacks.
- Process images with a Gaussian filter for smoothing, a Sato filter to highlight curvilinear structures, and a Hessian filter to generate binary images.
- Feature Extraction:
- Skeletonization: Convert binary images to 1-pixel-wide skeletons representing cytoskeletal fibers.
- Line Segment Features (LSFs): Extract metrics like fiber orientation (Orientational Order Parameter - OOP), length, and quantity.
- Cytoskeleton Network Features (CNFs): Analyze topology through graph networks, measuring connectivity, complexity, and radiality relative to the nucleus centroid.
Visualizing Cytoskeletal Signaling and Analysis Workflows
The following diagrams illustrate the key signaling pathways influenced by 3D microenvironments and the computational workflow for cytoskeletal analysis.
Mechanotransduction in 3D Microenvironments
Computational Cytoskeleton Analysis Pipeline
The Scientist's Toolkit: Essential Research Reagents
Successful cytoskeletal analysis relies on specific reagents and materials. The following table details key solutions for the protocols featured in this guide.
Table 2: Essential Research Reagents for Cytoskeletal Organization Studies
| Reagent/Material | Function/Application | Example from Literature |
|---|---|---|
| Type I Collagen (Rat Tail) | Major component of 3D hydrogels to mimic the native extracellular matrix (ECM). | Used for 3D fibroblast models in Leishmania infection studies and TFM [58] [56]. |
| Pluronics F127 | Non-ionic surfactant used to coat scaffolds and prevent non-specific cell adhesion. | Applied to 3D-printed PLA scaffolds in 3D muscle construct fabrication [59]. |
| Polyacrylamide (PA) Gels | Tunable, elastic substrates for 2D Traction Force Microscopy (TFM). | Used with a Young's Modulus of 5 kPa to mimic breast tumor stiffness [55]. |
| Blebbistatin | Specific inhibitor of myosin II, used to disrupt actomyosin contractility. | Applied at 50 µM to study the role of myosin in force generation [55]. |
| Nocodazole | Microtubule-depolymerizing agent used to investigate MT's role in cytoskeletal organization. | Used at 10 µM to perturb microtubule dynamics [55]. |
| Anti-α-Tubulin Antibody | Primary antibody for immunofluorescence staining of microtubule networks. | Essential for the computational pipeline analyzing microtubule architecture [1]. |
| Vibrational Spectroscopy (FT-IR) | Label-free, reagent-free method to analyze biomolecular changes in 2D vs. 3D cultures. | Used to identify lipid, protein, and nucleic acid alterations in prostate cancer stem cells [60]. |
The evidence overwhelmingly demonstrates that 3D culture models provide a superior platform for analyzing cytoskeletal organization in the context of cell invasion, minimizing the artifacts inherent to traditional 2D monolayers. The critical differences in traction forces, microtubule architecture, metabolic profiles, and resulting invasion strategies underscore the necessity of choosing a physiologically relevant model. While 2D systems retain utility for specific, reductionist questions, advancing our understanding of metastatic progression demands the adoption of 3D methodologies. By integrating advanced computational analysis, precise force measurements, and defined 3D matrices, researchers can effectively navigate the 2D vs. 3D conundrum, translating in vitro findings into clinically relevant insights with greater predictive power.
Optimizing Segmentation and Artifact Reduction in Dense or Confluent Cell Populations
Accurate segmentation of confluent cell populations is a critical yet challenging task in biomedical research, directly impacting the study of cytoskeletal organization and cell migration. This guide compares the performance of four advanced segmentation methodologies, providing researchers with objective data to select the optimal tool for their investigations into the phenotypic differences between invasive and non-invasive cells.
Performance Comparison of Segmentation Methods
The following table summarizes the key performance metrics of four distinct approaches, highlighting the trade-offs between accuracy, speed, and computational requirements.
Table 1: Performance Comparison of Cell Segmentation Methods
| Method Name | Core Approach | Reported Accuracy Metric | Reported Performance | Inference Speed | Primary Advantage |
|---|---|---|---|---|---|
| AKB-YOLO [61] | Deep Learning (YOLOv8-based with BiFPN & AKConv) | mAP50 (mask) | 95.0% | 38 FPS | Excellent accuracy & speed balance |
| Self-Supervised Learning (SSL) [62] | Pixel Classification (Optical Flow on original/blurred images) | F1 Score | 0.771 - 0.888 | Not Specified | No curated training data needed |
| Quantella [63] | Automated Image Analysis (Multi-exposure fusion & thresholding) | Deviation from Flow Cytometry | <5% | >10,000 cells/test | High-throughput, cost-effective |
| Image-based PAT Platform [64] [65] | Machine Learning (Pixel classification in cloud) | Not Specified | Validated for industrial use | Near-real-time | End-to-end automation, GMP-ready |
Detailed Experimental Protocols
To ensure reproducibility and provide context for the performance data, here are the detailed experimental protocols from the cited studies.
Table 2: Key Experimental Protocols and Cell Models
| Method | Cell Types / Tissues Used | Key Stains/Labels | Microscope & Imaging | Analysis Workflow |
|---|---|---|---|---|
| AKB-YOLO [61] | Glioma Stem Cells (GSCs) | Bright-field (unlabeled) | Inverted fluorescence microscope, 20x objective | CLAHE & Gaussian blur preprocessing → AKB-YOLO segmentation |
| SSL Approach [62] | MDA-MB-231, Hs27, S. cerevisiae | GFP, F-actin, Vinculin, DAPI | Phase contrast, DIC, epifluorescence, confocal (10x-63x) | Gaussian blur → Optical Flow calculation → Self-labeling → Pixel classification |
| Quantella [63] | CHO DG44, MCF-7, L929, RBCs | Trypan Blue | Smartphone-based platform with custom optics | Multi-exposure fusion → Thresholding → Morphological filtering |
| PAT Platform [64] [65] | Human Induced Pluripotent Stem Cells (hiPSCs) | Bright-field (label-free) | Evident CM20 incubation monitoring system | Automated image acquisition → Cloud processing → Pixel classification → Dashboard report |
AKB-YOLO for Glioma Stem Cells (GSCs)
This protocol is designed for complex, heterogeneous cell populations [61].
- Cell Culture & Imaging: Glioma stem cells (GSCs) were cultured in a serum-free neurosphere system. Time-lapse images were captured every 10 minutes for 4000 minutes using an inverted fluorescence microscope with a 20x objective, yielding 400 high-resolution (2048 x 2448 pixel) images [61].
- Image Preprocessing: A combination of Contrast-Limited Adaptive Histogram Equalization (CLAHE) and Gaussian blur was applied to balance noise suppression and local contrast enhancement, crucial for improving boundary definition [61].
- Model Training & Segmentation: The YOLOv8 baseline model was enhanced with a Bidirectional Feature Pyramid Network (BiFPN) for better multi-scale feature fusion and Adaptive Kernel Convolution (AKConv) for dynamic receptive field adjustment. The model was trained on manually annotated cell contours and used Probability Density-Guided Soft-NMS to reduce cell under-detection in dense regions [61].
Self-Supervised Learning (SSL) for High-Content Imaging
This method is ideal for scenarios where acquiring large, annotated training datasets is impractical [62].
- Imaging Diverse Modalities: Cells (e.g., MDA-MB-231, Hs27) were imaged using various modalities (phase contrast, DIC, epifluorescence) and magnifications (10x to 63x), with some samples stained for F-actin or vinculin [62].
- SSL Workflow: For each image, a Gaussian filter creates a blurred version. Optical flow vectors are then calculated between the original and blurred image pairs. These vectors serve as a self-labeled ground truth to train an image-specific pixel classifier for segmenting "cell" versus "background" without any pre-curated datasets [62].
- Phenotypic Quantification: The resulting high-fidelity segmentations enable the quantification of complex cellular features, such as membrane morphologies and organelle distribution, across different treatment conditions [62].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Cell Segmentation Experiments
| Item Name | Function / Application | Example Use Case |
|---|---|---|
| TrypLE Express [64] [65] | Dissociation of adherent cells for passaging or analysis. | Creating single-cell suspensions from hiPSC cultures. |
| ROCK Inhibitor (Y-27632) [64] [65] | Prevents apoptosis in dissociated cells, improving viability post-passaging. | Added to hiPSC cultures within 24 hours after seeding. |
| Recombinant Laminin 521 [64] [65] | A defined substrate for coating culture vessels to support cell adhesion and growth. | Coating CellSTACK and T-225 flasks for hiPSC culture. |
| Aggregated LDL (agLDL) [66] | Used to load vascular smooth muscle cells with lipids, modeling a disease state. | Studying cytoskeletal remodeling and migration in atherosclerosis research. |
| iC3b Complement Fragment [66] | Stimulates actin cytoskeleton reorganization and alters focal adhesion dynamics. | Investigating molecular signaling in lipid-loaded VSMC migration. |
| Trypan Blue [63] | A vital dye used to distinguish live (unstained) from dead (blue) cells. | Cell viability and density analysis in the Quantella platform. |
Method Selection Guide
Understanding the core principles of each method is key to making an informed choice. The following diagram illustrates the fundamental workflow of the AKB-YOLO and Self-Supervised Learning approaches.
The optimal segmentation method depends heavily on the research priorities. For most studies requiring high precision in analyzing cytoskeletal organization and dense populations in invasive vs. non-invasive cells, AKB-YOLO offers a powerful balance of high accuracy and speed [61]. In resource-limited settings or for novel cell types where annotated data is scarce, the SSL approach provides remarkable versatility without sacrificing performance [62]. For high-throughput industrial applications like therapy manufacturing, the automated PAT platform ensures robustness and compliance [64] [65], while Quantella presents a cost-effective alternative for routine viability and confluency checks [63]. This objective comparison empowers researchers to select the most effective tool, thereby enhancing the reliability of their findings in cell migration and cytoskeletal dynamics.
Cytoskeletal drugs, such as those targeting actin filaments and microtubules, are powerful tools in cell biology research and therapeutic development. While their primary intent is to specifically disrupt cytoskeletal dynamics, a growing body of evidence reveals that these compounds induce extensive off-target effects that significantly alter global cellular landscapes. These unintended consequences profoundly impact fundamental cellular processes, from protein folding and energy landscapes to metabolic regulation and cell detachment mechanics.
Understanding these pleiotropic effects is particularly crucial when comparing cytoskeletal organization in invasive versus non-invasive cells, as the very tools used to probe mechanisms can themselves alter the phenotypes under investigation. This guide systematically compares the off-target effects of major cytoskeletal drug classes, providing experimental data and methodologies essential for designing robust experiments and accurately interpreting results in cytoskeleton-focused research.
Mechanisms of Action and Off-Target Pathways
Primary and Secondary Mechanisms of Cytoskeletal Drugs
Table 1: Mechanisms of Action of Common Cytoskeletal Drugs
| Drug Name | Primary Target | Primary Effect | Key Off-Target Consequences | Cellular Functions Affected |
|---|---|---|---|---|
| Cytochalasin D | Actin filaments | Inhibits polymerization by capping barbed ends [67] | Alters macromolecular crowding; changes nonspecific surface interactions [68] | Protein folding stability; cytoplasmic fluidity |
| Latrunculin B | Actin filaments | Sequesters actin monomers [69] | Reduces cell adhesion; disrupts vinculin translocation [69] | Cell detachment; focal adhesion assembly |
| Nocodazole | Microtubules | Depolymerizes microtubules [68] | Decreases macromolecular crowding; alters protein energy landscapes [68] | Protein compactness; folding pathways |
| Vinblastine | Microtubules | Destabilizes microtubules [68] | Reduces cytoskeletal crowding despite decreasing cell volume [68] | Global protein stability and function |
| Blebbistatin | Myosin II | Inhibits non-muscle myosin II [70] | Disrupts actin-myosin cytoskeleton; affects drug reward behaviors [70] | Structural plasticity; motivation pathways |
Signaling Pathways Affected by Cytoskeletal Perturbation
The following diagram illustrates the key signaling pathways disrupted by cytoskeletal drugs, connecting primary targets to their downstream off-target effects:
Pathway Map: Cytoskeletal Drug Effects - This diagram maps how cytoskeletal drugs (yellow) initially target specific cytoskeletal components (red), leading to diverse off-target effects (green) that ultimately alter major cellular functions (blue).
Quantitative Comparison of Off-Target Effects
Experimental Evidence for Global Cellular Changes
Table 2: Experimentally Measured Off-Target Effects of Cytoskeletal Drugs
| Experimental System | Treatment | Measured Parameter | Quantitative Effect | Implications |
|---|---|---|---|---|
| Intracellular protein folding [68] | Cytochalasin D | PGK folding stability | Decreased compactness and folding stability | Altered energy landscapes for cytosolic proteins |
| Intracellular protein folding [68] | Latrunculin B | VlsE sensitivity | Opposite response to crowding vs sticking | Changed nonspecific surface interactions |
| Boyden chamber assay [69] | Latrunculin B | Cell detachment | Enhanced dissociation by abolishing actin fibers | Disrupted balance between anchoring and detachment |
| 3D in-gel spheroid [69] | TGF-β1 + Latrunculin B | Cell penetration | Increased detached cells from tumor spheres | Cytoskeleton maintains detachment balance |
| Viral replication [67] | Cytochalasin D | hMPV protein expression | 2 to 2.5 fold increase in fluorescent dots/cell | Actin disruption alters viral replication dynamics |
| In vitro actin polymerization [71] | ROS-induced severing | F-actin accumulation | Initial increase then disassembly with excess severing | Biphasic response to cytoskeletal disruption |
Methodologies for Assessing Off-Target Effects
Key Experimental Protocols
Protein Folding Landscape Analysis
Protocol 1: Measuring Cytoskeletal Drug Effects on Intracellular Protein Stability
Purpose: To quantify how cytoskeletal drugs alter protein folding energy landscapes inside living cells [68].
Reagents and Equipment:
- FRET-labeled proteins (PGK and VlsE) with different crowding sensitivities
- Cytoskeletal drugs: cytochalasin D, latrunculin B, nocodazole, or vinblastine
- Confocal fluorescence microscopy system with FRET capability
- Cell culture facilities and appropriate cell lines
- Environmental control for live-cell imaging
Procedure:
- Transfect cells with FRET-labeled sensor proteins (PGK as crowding sensor, VlsE as sticking sensor)
- Treat with cytoskeletal drugs targeting either actin (cytochalasin D, latrunculin B) or microtubules (nocodazole, vinblastine)
- Measure FRET efficiency changes over time using confocal microscopy
- Calculate folding stability and compactness from FRET ratios
- Validate crowding changes using dedicated crowding sensor CrH2-FRET
- Compare drug effects between different sensor proteins to distinguish crowding versus sticking effects
Key Measurements:
- PGK compaction changes indicate altered macromolecular crowding
- VlsE response patterns reveal changes in nonspecific surface interactions
- CrH2-FRET verification confirms overall crowding extent changes
Cell Detachment and Invasion Assessment
Protocol 2: 3D Spheroid Detachment Assay for Cytoskeletal Function
Purpose: To evaluate how cytoskeletal drugs disrupt cell adhesion and promote detachment in invasive cells [69].
Reagents and Equipment:
- Low-attachment 96-well U-plates for spheroid formation
- Matrigel or other extracellular matrix components
- Cytoskeletal drugs: latrunculin B, TGF-β1 neutralizing antibodies
- Immunofluorescence staining reagents for actin and vinculin
- Confocal or high-content imaging system
Procedure:
- Form tumor spheroids by culturing cells in U-plates for 5-7 days using low-speed centrifugation
- Embed spheroids in Matrigel and allow polymerization for 24-48 hours
- Treat with cytoskeletal drugs (e.g., latrunculin B at determined IC50 concentrations)
- For TGF-β inhibition studies, include TGF-β1 neutralizing antibody treatments
- Image spheroids over time using confocal microscopy
- Quantify detached cells by integrating two-dimensional invasion sections in the perimembrane area
- Co-stain for actin fibers and vinculin to assess focal adhesion disruption
Key Measurements:
- Number of detached cells from main spheroid body
- Distance of cell penetration into Matrigel
- Actin fiber integrity and vinculin translocation patterns
- Comparison between drug-treated and control conditions
The following diagram illustrates the experimental workflow for assessing cytoskeletal drug effects using these key methodologies:
Experimental Workflow for Assessing Off-Target Effects - This diagram outlines two key methodological approaches (red) for characterizing cytoskeletal drug effects: protein folding assays (green) and 3D spheroid detachment protocols (blue), leading to comprehensive off-target effect characterization.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Cytoskeletal Off-Target Effect Studies
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Actin-Targeting Drugs | Cytochalasin D, Latrunculin B | Disrupt actin polymerization; study cell adhesion, protein folding | Different mechanisms: capping vs. monomer sequestration |
| Microtubule-Targeting Drugs | Nocodazole, Vinblastine | Depolymerize microtubules; examine intracellular transport | Consider cell type-specific sensitivity and exposure time |
| FRET-Based Sensors | PGK-FRET, VlsE-FRET, CrH2-FRET | Monitor protein folding landscapes and crowding changes | Requires appropriate controls for FRET efficiency calculations |
| 3D Culture Systems | Matrigel, U-plate spheroid formation | Model tumor microenvironment and invasive cell behavior | Matrix composition significantly affects results |
| Signaling Modulators | TGF-β1 neutralizing antibodies, Rho GTPase modulators | Probe specific pathways in cytoskeletal regulation | Timing critical for pathway-specific interventions |
| Visualization Tools | Phalloidin (F-actin), tubulin antibodies, vinculin markers | Cytoskeletal and focal adhesion imaging | Fixation methods affect cytoskeletal preservation |
Implications for Invasive vs. Non-Invasive Cell Research
The off-target effects of cytoskeletal drugs present both challenges and opportunities when studying differences between invasive and non-invasive cells. In invasive cells, which typically exhibit enhanced cytoskeletal dynamics and mechanical adaptation capabilities, cytoskeletal drugs can produce markedly different off-target effects compared to their non-invasive counterparts.
Research demonstrates that TGF-β1-mediated vimentin expression and focal adhesion assembly create a dependency on actin cytoskeleton remodeling for maintaining the balance between cell anchoring and detachment [69]. When this balance is disrupted by drugs like latrunculin B, invasive leading cells exhibit enhanced detachment—an effect not observed in non-invasive cells. This differential response highlights how the same cytoskeletal drug can produce fundamentally different outcomes depending on the cellular context and underlying cytoskeletal organization.
Furthermore, the protein folding consequences of cytoskeletal disruption may disproportionately affect invasive cells, which often operate under metabolic and proteostatic stress. The observation that cytoskeletal drugs alter macromolecular crowding [68] suggests that invasive cells relying on precise folding of metastatic proteins may be particularly vulnerable to these off-target effects.
The evidence presented in this guide demonstrates that cytoskeletal drugs induce significant off-target effects that extend far beyond their intended targets, globally altering cellular landscapes through changes in macromolecular crowding, surface interactions, protein folding environments, and adhesion dynamics. When comparing cytoskeletal organization in invasive versus non-invasive cells, researchers must account for these pleiotropic effects in their experimental designs.
Best practices for future research include:
- Employing multiple complementary assessment methods (e.g., FRET-based sensors with 3D culture models)
- Including appropriate controls for off-target effects in experimental design
- Considering temporal aspects of drug exposure, as acute versus chronic treatments produce different adaptive responses
- Acknowledging cell-type specific variations in sensitivity to cytoskeletal perturbations
- Validating findings using multiple cytoskeletal drugs with similar intended targets but different mechanisms
By systematically accounting for these off-target effects, researchers can more accurately interpret experimental results and advance our understanding of cytoskeletal differences between invasive and non-invasive cells, ultimately contributing to more effective therapeutic strategies targeting the cytoskeleton in disease contexts.
The selection of appropriate cell line models is a critical foundation for biomedical research, particularly in studies of cytoskeletal organization and cellular invasion. While cell lines offer a reproducible and accessible system for investigating cancer biology, their genetic and phenotypic fidelity to the original tumors varies considerably. A growing body of evidence demonstrates that inadequate attention to genetic background and tissue origin during model selection can compromise research reproducibility and clinical translatability. This guide provides a systematic framework for evaluating cell line models, with special emphasis on accounting for genomic divergence and origin-specific characteristics that influence cytoskeletal dynamics and invasive behavior.
Genomic Landscape of Common Cell Line Models
Comprehensive genomic comparisons between cell lines and primary tumor samples have revealed substantial differences that researchers must consider when selecting models for their investigations.
Table 1: Genomic Comparison of Cell Lines and Primary Tumors
| Characteristic | HGSOC Tumors (TCGA) | Ovarian Cancer Cell Lines (CCLE) | Breast Cancer Tumors (TCGA) | Breast Cancer Cell Lines (CCLE) |
|---|---|---|---|---|
| Median fraction of genome altered | 46% | Variable, wider distribution | Profile-dependent | Profile-dependent |
| TP53 mutation prevalence | 95% | 62% | Subtype-dependent | Subtype-dependent |
| Typical mutational frequency | 1.6 per Mb | 4.3 per Mb | Variable | Variable |
| BRCA1/2 mutation prevalence | >10% | 6-9% | Hereditary in 5-10% | Variable |
Analysis of high-grade serous ovarian cancer (HGSOC) models reveals that commonly used ovarian cancer cell lines such as SK-OV-3, A2780, OVCAR-3, CAOV3, and IGROV1 show pronounced molecular differences from primary HGSOC tumors [72]. Notably, five ovarian cancer cell lines—IGROV1, OC316, EFO27, OVK18, and TOV21G—exhibit a distinctive "hypermutator" genotype characterized by surprisingly high mutation counts despite few copy-number alterations, setting them clearly apart from most HGSOC tissue samples [72].
Similarly, studies of breast cancer models demonstrate that MCF-7 cells, while extensively used, show only minimal similarity in biological processes with human breast cancer tissues, with fundamental functions like cell cycle being among the few conserved features [73]. Network topology metrics reveal drastic differences in gene behavior between MCF-7 and human breast cancer tissues, suggesting that reliance on this single model may cause researchers to miss important therapeutic targets [73].
Impact of Genetic Divergence on Cytoskeletal Organization and Invasive Behavior
The cytoskeleton undergoes dramatic reorganization during cancer progression, and genetic background significantly influences these architectural changes that underlie invasive potential.
Table 2: Cytoskeletal Features Associated with Invasive Potential
| Cytoskeletal Component | Feature | Non-Invasive/Epithelial-like | Invasive/Mesenchymal-like |
|---|---|---|---|
| Microtubules | Organization | Well-organized, radial | Disorganized, disperse orientations |
| Length | Longer filaments | Shorter filaments | |
| Distribution | Sparsely distributed | Compact distribution | |
| Actin Filaments | Architecture | Cortical actin arcs | Prominent stress fibers |
| Membrane protrusions | Limited | Extensive lamellipodia/filopodia | |
| Perinuclear actin cap | Often present | Frequently absent | |
| Focal Adhesions | Size | Variable | Larger in some invasive types |
| Distribution | Cell periphery | Ends of stress fibers |
Research on malignant mesothelioma (MM) cell lines demonstrates that highly malignant and invasive MM cells possess distinct organization of both the actin filament and vimentin systems compared to less malignant MM cell lines [74]. In breast cancer models, cells with disrupted E-cadherin and increased invasive potential display markedly different cytoskeletal architecture, with microtubules that are shorter, have disperse orientations, and are more compactly distributed [1].
A novel computational pipeline for characterizing cytoskeletal architecture has identified multiple quantitative metrics that distinguish invasive cells, including microtubule orientation, compactness, radiality, and morphology [1]. This approach enables researchers to move beyond traditional qualitative assessments to precise quantification of cytoskeletal features predictive of invasive behavior.
Experimental Protocols for Model Validation
Genomic Concordance Assessment
Purpose: To evaluate the molecular similarity between candidate cell lines and the primary tumor type they are intended to model.
Methodology:
- Obtain genomic data (copy number alteration, mutation, mRNA expression) from resources like TCGA for tumors and CCLE for cell lines.
- Calculate the correlation of CNA profiles between each cell line and the mean CNA profile of tumors.
- Assess alterations in key cancer genes characteristic of the tumor subtype.
- For breast cancer models, apply PAM50 gene expression subtyping to both tumors and cell lines to verify concordance of molecular subtypes [75].
- Generate a composite similarity score integrating multiple genomic features to rank cell lines by their suitability as models [75].
Interpretation: Cell lines with higher correlation scores and characteristic subtype-specific alterations are preferable for studies requiring high clinical relevance.
Cytoskeletal Phenotyping Pipeline
Purpose: To quantitatively characterize cytoskeletal organization and its relationship to invasive potential.
Methodology:
- Culture cells on appropriate extracellular matrix substrates (e.g., laminin) to mimic physiological conditions [1].
- Fix and stain for cytoskeletal components: α-tubulin for microtubules, phalloidin for F-actin, and antibodies for vimentin (intermediate filaments) [74].
- Acquire high-resolution z-stack images using fluorescence microscopy.
- Process images through deconvolution to remove noise and blur, improving contrast and resolution [1].
- Apply curvilinear structure enhancement filters (Sato filter) and generate binary images of cytoskeletal structures [1].
- Skeletonize binary images to enable extraction of quantitative parameters:
- Line Segment Features (LSFs): Orientation, length, bundling
- Cytoskeleton Network Features (CNFs): Connectivity, complexity, radiality [1]
- Calculate specific metrics including Orientational Order Parameter (OOP) for alignment, number of fibers per cell area for compactness, and radial score relative to nucleus centroid [1].
Interpretation: Compare these quantitative metrics between experimental conditions or across cell lines with different genetic backgrounds to establish correlations between cytoskeletal organization and invasive behavior.
Cell Line Evolution and Its Implications
Cancer cell lines are not genetically stable entities but evolve in laboratory conditions, acquiring changes that affect their responses to drugs and likely their cytoskeletal organization [76]. Deep molecular analyses of multiple strains of widely used lines like MCF-7 (breast cancer) and A549 (lung cancer) have revealed striking genomic differences ranging from single base pair mutations to large-scale structural variations and significant gene expression differences [76].
This evolution is influenced by laboratory conditions, including the nutrient media composition, which can give some cells within a strain a growth advantage, allowing genetically distinct populations to emerge [76]. These genetic changes directly impact drug sensitivity; for instance, MCF-7 strains lacking estrogen receptor expression show reduced sensitivity to tamoxifen, correlating with their genetic profiles [76].
Visualization of Relationships and Workflows
Relationship Between Cell Line Properties and Research Outcomes
Diagram Title: Factors Influencing Cell Line Model Relevance
Experimental Workflow for Cytoskeletal Analysis
Diagram Title: Cytoskeletal Analysis Computational Pipeline
Research Reagent Solutions
Table 3: Essential Research Reagents for Cell Line Characterization
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cell Line Collections | CCLE, ATCC, Horizon Discovery | Source of genetically characterized cell lines |
| Gene Editing Tools | CRISPR-Cas9, rAAV | Engineering specific genetic alterations |
| Cytoskeletal Stains | α-tubulin antibodies, Phalloidin (F-actin), Vimentin antibodies | Visualizing cytoskeletal components |
| Image Analysis Tools | Custom computational pipeline, ImageJ, Commercial software | Quantifying cytoskeletal features |
| Extracellular Matrix | Laminin, Collagen, Fibronectin | Mimicking physiological microenvironment |
| Microscopy Systems | Fluorescence microscope with z-stack capability | High-resolution imaging of cellular structures |
Strategic selection of cell line models requires careful consideration of genetic background, tissue origin, and intended research applications. The genomic divergence between commonly used cell lines and primary tumors necessitates thorough molecular characterization before initiating studies, particularly for cytoskeletal organization and invasion research where subtle phenotypic differences can significantly impact findings. By implementing the validation protocols and quantitative approaches outlined in this guide, researchers can enhance the reliability and clinical relevance of their cell line-based investigations, ultimately advancing our understanding of the cytoskeletal mechanisms underlying cancer progression.
High-sensitivity motility assays are indispensable tools in modern cell biology and drug discovery, particularly for research focused on cytoskeletal organization and the invasive potential of cancer cells. These assays enable researchers to dissect the intricate relationship between the cytoskeleton's architecture and cell motility, a critical determinant in processes like cancer metastasis. However, a significant challenge persists: the variance inherent in these sensitive techniques can compromise the reliability and reproducibility of data. This guide objectively compares emerging technologies and methodologies that aim to standardize motility analysis. We will evaluate advanced platforms for single-cell tracking, innovative computational pipelines for cytoskeletal analysis, and machine learning-enhanced tools, providing a clear comparison of their performance in mitigating assay variance and yielding precise, actionable data.
Technology Performance Comparison
The following table summarizes the core performance metrics of three advanced approaches for analyzing cell motility and cytoskeletal organization, providing a direct comparison of their capabilities in standardizing assays.
Table 1: Comparative Performance of High-Sensitivity Motility and Cytoskeletal Assays
| Assay Technology | Key Measured Parameters | Reported Sensitivity/Specificity/Accuracy | Key Advantages for Standardization |
|---|---|---|---|
| Nanowell-in-Microwell Single-Cell Motility Assay [77] | Cell motility (µm/h), Elongation rate, Motility phenotypes (Type I-III) | Motility: 4.4 ± 3.1 µm/h (0% FBS), 12.2 ± 9.6 µm/h (10% FBS), 15.9 ± 11.0 µm/h (TNF-α) [77] | Isolates single cells to eliminate cell-cell interaction variance; enables high-throughput profiling. |
| Computational Cytoskeletal Architecture Pipeline [1] | Microtubule orientation (OOP), quantity (Nl), compactness (Nl/Ac), radiality (RS), fiber-nucleus distance (Di) | Distinguished mutant E-cadherin invasive cells by significantly lower OOP, shorter, disoriented microtubules [1] | Provides multiple quantitative, objective metrics from standard immunofluorescence images, reducing subjective bias. |
| Machine Learning (ATLAS) for IVMA [78] | Actin filament velocity, Actin filament length | Accurately measures velocity and length across a broad range of experimental conditions [78] | Removes human bias from filament tracking; fast, efficient analysis of high-throughput data. |
Detailed Experimental Protocols
To ensure reproducibility, this section outlines the core methodologies for the key technologies compared.
Protocol: High-Throughput Single-Cell Motility Assay
This protocol details the use of nanowell-in-microwell plates for analyzing single-cell motility, a method that effectively isolates cells to minimize environmental variance [77].
- Step 1: Platform Fabrication. Fabricate open-top nanowells (e.g., 70 × 70 × 60 μm) arranged in high-density arrays on a glass slide using photolithography. Bond the patterned slide to a standard 384-well plastic plate frame, resulting in approximately 1200 nanowells per microwell [77].
- Step 2: Cell Seeding and Preparation. Seed cells into each microwell at a density corresponding to roughly 30% of the total number of nanowells. This density maximizes single-cell occupancy based on Poisson statistics. Culture the cells for 48 hours to promote adhesion and acclimatization to the substrate [77].
- Step 3: Fluorescent Labeling and Imaging. Fluorescently label live cells using a viability marker, such as Calcein Green. Use an automated microscope to capture time-lapse images of each nanowell at regular intervals (e.g., every hour) over a defined period (e.g., 12 hours) [77].
- Step 4: Image Analysis and Tracking. Employ a custom analysis pipeline:
- Segmentation: Apply a 2D grid to brightfield images to define the coordinates of individual nanowells.
- Filtering: Filter out invalid nanowells (those without a live cell, with more than one cell, or where cell division occurs during imaging).
- Tracking: For each valid nanowell, extract the centroid position and cell length (longest axis) from each time-lapse frame. Calculate average motility (µm/h) and elongation rate [77].
Protocol: Computational Analysis of Cytoskeletal Architecture
This protocol describes an image-based pipeline to quantify cytoskeletal organization from standard immunofluorescence images, providing objective metrics to replace subjective assessment [1].
- Step 1: Sample Preparation and Imaging. Culture cells on coverslips under the required experimental conditions. Fix cells and perform immunofluorescence staining for cytoskeletal components of interest (e.g., α-tubulin for microtubules). Acquire multiple images along the Z-axis for each channel using a fluorescence microscope [1].
- Step 2: Image Pre-processing. Perform maximum intensity projection (MIP) on the Z-stacks to create 2D images. Subject these images to deconvolution to remove noise and blur, improving contrast and resolution. Subsequently, process images with a Gaussian filter to smooth the fluorescence signal and a Sato filter to highlight curvilinear structures [1].
- Step 3: Fiber Segmentation and Skeletonization. Generate binary images from the processed images using a Hessian filter. Skeletonize the binary images to create a simplified one-pixel-wide representation of the cytoskeletal fibers [1].
- Step 4: Feature Extraction. Analyze the skeletonized images using two strategies:
- Line Segment Features (LSFs): Extract metrics like fiber orientation (Orientational Order Parameter - OOP), length, and quantity.
- Cytoskeleton Network Features (CNFs): Extract metrics based on graph networks, such as compactness (fibers per cell area), radiality (radial score relative to the nucleus), and fiber-nucleus interconnection (average distance) [1].
Signaling Pathways and Workflow Diagrams
The following diagrams illustrate the logical workflow of the two primary experimental protocols discussed, providing a visual guide to their standardized processes.
Diagram 1: Workflows for single-cell motility and cytoskeletal analysis.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of high-sensitivity, standardized assays relies on a core set of reagents and materials. The table below lists essential items for the featured experiments.
Table 2: Essential Research Reagents and Materials for Motility and Cytoskeletal Assays
| Item Name | Function/Application | Specific Example from Research |
|---|---|---|
| Nanowell-in-Microwell Plates | Provides a high-throughput platform for physically confining and tracking individual cells. | Custom-fabricated plates with 70x70x60 µm nanowells in a 384-well format [77]. |
| Calcein Green AM / Calcein AM | Fluorescent viability dye used for live-cell staining and tracking. | Used to fluorescently label live MDA-MB-231 cells for motility tracking in nanowells [77]. |
| α-Tubulin Antibody | Primary antibody for immunofluorescence staining of microtubule networks. | Used to stain the microtubule component of the cytoskeleton for computational analysis [1]. |
| TRITC-conjugated Phalloidin | High-affinity fluorescent probe used to stain and visualize filamentous actin (F-actin). | Used to visualize F-actin structures (stress fibers, lamellipodia) in malignant mesothelioma cell lines [74]. |
| Functional Lumen Imaging Probe (FLIP) | Measures esophageal diameter and contractility; complements manometry for motility disorders. | Used alongside manometry and timed barium esophagram to diagnose esophageal motility disorders [79]. |
| Selenite Broth (SB) | Selective enrichment broth used for the culture of Salmonella from stool samples. | Used as a selective enrichment step in stool culture for diagnosing Salmonella enterica [80]. |
Discussion and Comparative Analysis
The comparative data reveals that the fundamental strategy for mitigating variance lies in leveraging physical confinement and computational objectivity. The Nanowell-in-Microwell Assay addresses cellular heterogeneity—a major source of variance in bulk assays—by providing isolated, single-cell microenvironments. This allows for the clear identification of subpopulations, such as the highly motile cells that persist even under therapeutic challenge [77]. The technology's strength is its direct measurement of cell behavior, providing robust, quantitative motility data (e.g., 12.2 ± 9.6 µm/h under standard culture) that is free from the confounding effects of cell-cell interactions.
In parallel, the Computational Cytoskeletal Pipeline tackles the variance introduced by subjective human interpretation. By extracting multiple, quantitative descriptors (OOP, compactness, radiality) from standard images, it transforms qualitative observations into a rich, objective dataset [1]. This method has proven sensitive enough to detect the specific cytoskeletal signature of invasive cells, such as the disorganized microtubule networks in E-cadherin mutant cells. This approach is highly complementary to motility assays, as it provides the underlying structural explanation for the observed behavioral phenotypes.
Emerging tools like ATLAS, which uses machine learning to track actin filament motility, represent the next logical step in standardization [78]. By fully automating the analysis of dynamic processes, these systems remove human bias and increase throughput, further enhancing reproducibility. Together, these technologies provide researchers with a powerful, multi-faceted toolkit to obtain sensitive, reliable, and standardized data on cell motility and its cytoskeletal foundations, thereby accelerating research in cancer biology and drug development.
Benchmarking and Therapeutic Translation: Validating Cytoskeletal Targets and Signatures
The metastatic cascade represents the primary cause of cancer-related mortality, necessitating advanced understanding of its underlying cellular mechanisms. Central to this process is the cytoskeleton, a dynamic network of structural filaments that dictates cell shape, mechanical properties, and migratory capabilities. This guide provides a comprehensive comparative analysis of specific cytoskeletal alterations correlated with invasive potential, organizing quantitative data and experimental methodologies to facilitate research in cancer cell biology and drug development. By synthesizing data from multiple studies across cancer types, we establish a framework for identifying cytoskeletal biomarkers of invasion and metastasis.
Quantitative Cytoskeletal Feature Comparison in Invasive vs. Non-Invasive Cells
Research across multiple cancer types reveals consistent patterns of cytoskeletal reorganization correlated with invasive potential. The tables below synthesize quantitative findings from comparative studies of invasive and non-invasive cancer cells.
Table 1: Microtubule Organization Features in Invasive Cells
| Cytoskeletal Feature | Non-Invasive Cells | Invasive Cells | Measurement Method | Biological Significance |
|---|---|---|---|---|
| Orientational Order Parameter (OOP) | Higher values (e.g., ~0.475) [1] | Significantly lower values [1] | Angular distribution (θi) of cytoskeleton [1] | Indicates fiber alignment; lower OOP = dispersed orientations |
| Microtubule Length | Longer fibers [1] | Shorter fibers [1] | Line Segment Features (LiE) [1] | Shorter fibers associate with disrupted E-cadherin |
| Fiber Compactness | More dispersed distribution (e.g., 0.421 μm⁻²) [1] | More compactly distributed (e.g., 2.039 μm⁻²) [1] | Number of fibers per cell area (Nl/Ac) [1] | Compactness may reflect cytoskeletal condensation |
| Radial Distribution | Variable radial patterns [1] | Disrupted radiality relative to nucleus [1] | Radial score (RS) relative to nucleus centroid [1] | Reflects organization from cell center |
Table 2: Actin and Vimentin Organization in Mesothelioma Cell Lines of varying Malignancy
| Cytoskeletal Feature | Less Malignant/Epithelioid | Highly Malignant/Sarcomatoid | Measurement Method | Biological Significance |
|---|---|---|---|---|
| Cell Morphology | Small, round, aspect ratio ~1.6 [74] | Elongated, fibroblast-like, aspect ratio ~3.6 [74] | Microscopy & morphological analysis [74] | Elongation indicates mesenchymal transition |
| F-actin Organization | Cortical actin arcs, broad lamellipodia [74] | Prominent stress fibers, small lamellipodia [74] | TRITC-phalloidin staining [74] | Stress fibers enable contractile forces |
| Perinuclear Actin Cap | Often absent [74] | Present in many cells (except most invasive) [74] | Fluorescence microscopy [74] | Correlates with differentiated phenotype |
| Focal Adhesion Size | Relatively uniform (~1.15-1.20 μm²) [74] | Variable: larger (1.49 μm²) or smaller (0.91 μm²) [74] | Anti-phosphotyrosine staining [74] | Altered adhesion signaling in invasion |
| Vimentin Organization | Distinct from invasive pattern [74] | Distinct organization associated with invasiveness [74] | Immunofluorescence [74] | Mesenchymal marker overexpression |
Table 3: Mechanical Properties and Gene Expression Correlations
| Parameter | Epithelial/ Less Metastatic | Mesenchymal/ Highly Metastatic | Correlation with Invasion | Measurement Technique |
|---|---|---|---|---|
| Young's Modulus | Stiffer (e.g., OVCAR3) [81] | Softer (e.g., HEY, HEY A8) [81] | Inverse correlation with metastatic potential [81] | Atomic Force Microscopy [81] |
| Viscous Time Constant | Reduced (slower relaxation) [81] | Increased (faster relaxation) [81] | Associated with fluid cellular state [81] | Zener model of viscoelasticity [81] |
| Metastatic Gene Correlation | Lower correlation with mechanics [81] | Higher correlation with mechanics [81] | Mechanics linked to metastatic programs [81] | Single-cell RT-qPCR [81] |
Experimental Protocols for Cytoskeletal Analysis
Computational Pipeline for Microtubule Architecture Dissection
A novel bioimaging pipeline enables quantitative characterization of cytoskeletal fiber organization from immunofluorescence images [1]:
Sample Preparation and Imaging:
- Culture cells on laminin-coated surfaces to provide supportive growth environment
- Perform immunofluorescence staining for α-tubulin and nucleus
- Acquire multiple Z-stack images for each channel
Image Preprocessing:
- Apply deconvolution to remove noise and blur, improving contrast and resolution
- Perform maximum intensity projection (MIP) of Z-stacks to create 2D images
- Process with Gaussian filter to smooth fluorescence signal
- Apply Sato filter to highlight curvilinear structures
- Use Hessian filter to generate binary images
- Skeletonize binary images to enable cytoskeletal parameter calculation
Feature Extraction:
- Calculate Line Segment Features (LSFs): orientation, morphology, quantity
- Determine Cytoskeleton Network Features (CNFs): compactness, radiality, connectivity
- Perform automatic nuclei segmentation to define nuclei centroids and area
- Extract nuclei-cytoskeletal ratios and spatial relationships
This pipeline successfully distinguished microtubule signatures of cells expressing mutant E-cadherin (associated with invasive phenotype) from wild-type cells [1].
Actin Organization and Focal Adhesion Analysis Protocol
For comprehensive actin cytoskeleton assessment in malignant mesothelioma cell lines [74]:
Cell Culture and Staining:
- Seed cells on coverslips and culture for 24 hours
- Fix cells and permeabilize using standard protocols
- Stain with TRITC-conjugated phalloidin to visualize F-actin
- Counterstain with anti-phosphotyrosine antibody (PY99) to identify focal adhesions
- Include nuclear staining for reference
Microscopy and Qualitative Assessment:
- Examine by fluorescence microscopy for stress fibers, actin arcs, and lamellipodia
- Classify cells based on dominant actin structures
- Assess at least 100 cells per condition across multiple experiments
Quantitative Analysis:
- Measure focal adhesion size using ImageJ software
- Determine cell aspect ratio and circularity through elliptical fitting
- Evaluate presence or absence of perinuclear actin caps
- Categorize cells based on histological subtype (epithelioid, sarcomatoid, biphasic)
Integrated Mechanical Properties and Gene Expression Analysis
The single cell "genomechanics" method correlates mechanical properties with molecular profiles [81]:
Sample Preparation:
- Culture cells on gridded coverslips for cell tracking
- Use three ovarian cancer cell lines of varying metastatic potential (OVCAR3, HEY, HEY A8)
Mechanical and Morphological Measurements:
- Acquire optical images for morphological analysis (size, aspect ratio, circularity, solidity)
- Perform AFM indentation force curves to measure Young's modulus
- Calculate viscous relaxation time constants using Zener model of viscoelasticity
Single Cell Isolation and Gene Expression:
- Locate cells using gridded substrate as registry
- Isolate individual cells via micropipette aspiration
- Deposit cells into tubes for immediate lysis
- Perform RT-qPCR using Fluidigm BioMark 96.96 Dynamic Array
- Analyze 96 genes related to EMT, metastatic enhancers/suppressors, cancer stemness, ECM remodeling, and cytoskeletal remodeling
This integrated approach revealed that cell stiffness correlates more highly with metastatic programs than with cytoskeletal genes alone [81].
Signaling Pathways Connecting Cytoskeleton to Invasion
The relationship between cytoskeletal reorganization and invasive capacity is governed by specific molecular pathways. The diagram below illustrates key regulatory mechanisms:
Cytoskeletal Regulation of Invasion Pathways
Key pathway components include:
- TGF-β1 Signaling: External mechanical forces activate TGF-β1, which stimulates actin polymerization through the RhoA-LIMK2-cofilin pathway and upregulates vimentin expression [69].
- Vimentin-Focal Adhesion Crosstalk: Vimentin controls focal adhesion kinase (FAK) activity through the VAV2-Rac1 pathway, orchestrating focal adhesion complex assembly [69].
- Mechanical Transduction: Matrix rigidity enhances TGF-β1 activation, creating a feedback loop that promotes cytoskeletal reorganization toward invasive phenotypes [82] [69].
- Dynamic Reciprocity: Migratory cells continuously adjust their mechanical properties in response to physical changes in the extracellular matrix, enabling migratory plasticity [82].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Cytoskeletal-Invasion Correlation Studies
| Reagent/Category | Specific Examples | Research Application | Functional Role |
|---|---|---|---|
| Cytoskeletal Stains | TRITC-phalloidin [74], α-tubulin antibodies [1] [74] | Fluorescence visualization of cytoskeletal structures | Selective binding to F-actin or tubulin for architecture analysis |
| Focal Adhesion Markers | Anti-phosphotyrosine (PY99) [74] [83], anti-vinculin [69] | Identification of adhesion complexes | Labels focal adhesions and attachment points |
| Inhibitors/Tools | Imipramine Blue [83], Latrunculin B [69], Honokiol [83] | Experimental manipulation of cytoskeleton | Targets ROS production or directly disrupts actin polymerization |
| Invasion Assay Systems | Boyden chambers [69], 3D in-gel spheroids [69], Transwell inserts [84] | Quantitative invasion measurement | Models different aspects of metastatic invasion |
| Mechanical Profiling | Atomic Force Microscopy [81], Micropipette aspiration [81] | Single-cell biomechanical properties | Measures stiffness, viscoelasticity correlated with invasion |
| Gene Expression Analysis | Multiplexed RT-qPCR [81], Fluidigm BioMark dynamic arrays [81] | Molecular profiling of single cells | Correlates cytoskeletal features with metastatic gene programs |
Comparative Analysis of Research Models
Table 5: Experimental Models for Cytoskeletal-Invasion Studies
| Model System | Applications | Key Cytoskeletal Features Analyzable | Technical Considerations |
|---|---|---|---|
| E-cadherin Mutant Models [1] | Study loss of cell-cell adhesion and invasion | Microtubule organization parameters (OOP, length, compactness) | Requires genetic manipulation; specific to epithelial-derived cancers |
| Mesothelioma Cell Panel [74] | Correlation with histological subtypes | Actin organization, vimentin, focal adhesion size | Captures spectrum of epithelial to sarcomatoid transition |
| 3D in-gel Spheroid Assays [69] | Analysis of cell detachment and collective invasion | Actin fiber formation, vinculin translocation | More physiologically relevant; technically challenging imaging |
| Ovarian Cancer Metastatic Series [81] | Linking mechanics, gene expression and invasion | Whole-cell mechanical properties, morphological parameters | Enables integrated multi-parameter analysis at single-cell level |
| Glioblastoma PTEN/p53 Models [85] | Genetic determinant studies of invasion | Actin distribution, invasion capacity through matrices | Isolates specific genetic contributions to cytoskeletal regulation |
This comparative guide demonstrates that invasive potential across cancer types correlates with measurable cytoskeletal alterations including microtubule disorganization, specific actin structures, vimentin reorganization, and reduced cellular stiffness. The consistent patterns emerging across diverse models provide researchers with validated biomarkers and methodologies for assessing metastatic potential and screening therapeutic compounds targeting the invasion machinery.
The cytoskeleton, a complex and dynamic network of protein filaments, is fundamental to cellular architecture and function. Its role is particularly pivotal in cancer biology, where the reprogramming of cytoskeletal organization is a hallmark of invasive and metastatic cells. Among its core components, microtubules are crucial for maintaining cell shape, enabling intracellular transport, and, most critically, orchestrating cell division through the formation of the mitotic spindle [86] [87]. Microtubule-targeting agents (MTAs) represent a cornerstone of cancer chemotherapy, leveraging this essential cellular machinery to halt the proliferation of cancer cells [86] [87]. These compounds are traditionally classified into two broad categories based on their primary effect on microtubule dynamics: stabilizers and destabilizers. However, their efficacy and toxicity profiles vary significantly between different agents, driven by their distinct binding sites on the tubulin heterodimer and their unique pharmacokinetic properties [86] [87]. This guide provides a comparative profiling of major MTAs, framing their mechanisms and performance within the broader context of cytoskeletal research in cancer. It is designed to equip researchers and drug development professionals with structured experimental data and protocols to inform future investigative and therapeutic work.
Comparative Profiling of Major Microtubule-Targeting Agents
Mechanism of Action and Binding Site Classification
Microtubules are cylindrical polymers composed of αβ-tubulin heterodymers, whose dynamic growth and shrinkage—a property known as dynamic instability—are governed by GTP hydrolysis [87]. MTAs interfere with this precise dynamics, ultimately triggering cell cycle arrest and apoptosis [87]. The therapeutic effects of these agents are mediated through interactions with specific binding sites on the tubulin protein. Structural biology and drug discovery efforts have now mapped at least nine distinct binding pockets, offering diverse pharmacological entry points [86]. The seven well-established sites include the taxane, vinca alkaloid, colchicine, maytansine, laulimalide/peloruside A, pironetin, and the recently discovered gatorbulin binding sites [87].
Table 1: Classification and Mechanisms of Microtubule-Targeting Agents
| Agent Class | Representative Drugs | Primary Mechanism | Binding Site | Cellular Outcome |
|---|---|---|---|---|
| Microtubule Stabilizers | Paclitaxel, Docetaxel, Cabazitaxel, Ixabepilone [87] | Promotes microtubule assembly and stability; inhibits depolymerization [87] | Taxane site (on β-tubulin) [87] | Cell cycle arrest at G2/M phase, disrupted mitotic spindle formation, apoptosis [87] |
| Microtubule Destabilizers | Vinblastine, Vincristine, Vinorelbine [87] | Inhibits tubulin polymerization, leading to microtubule shortening [87] | Vinca alkaloid site (on β-tubulin) [87] | Suppression of microtubule dynamics, mitotic spindle disruption, apoptosis [87] |
| Microtubule Destabilizers | Combretastatin A-4 [87] | Inhibits tubulin polymerization [87] | Colchicine site (on β-tubulin) [87] | Disruption of mitotic spindle, apoptosis, and vascular disruption in tumors [87] |
The following diagram illustrates the mechanism of action of these major MTA classes on microtubule dynamics during mitosis.
Diagram 1: Mechanism of microtubule-targeting agents in disrupting cell division.
Comparative Efficacy and Safety in Clinical Applications
The clinical utility of different MTAs varies across cancer types and treatment lines, influenced by factors such as efficacy, resistance mechanisms, and toxicity profiles. Direct and indirect comparisons in clinical trials provide critical insights for therapy selection.
Table 2: Comparative Clinical Efficacy of Taxanes in Specific Cancers
| Cancer Type | Therapeutic Context | Compared Regimens | Key Efficacy Findings | Citation |
|---|---|---|---|---|
| Esophageal Cancer | Second-line chemotherapy | Docetaxel (DTX) vs. Weekly Paclitaxel (wPTX) | wPTX showed superior disease control rate (64.3% vs 28%, P=0.043) and progression-free survival (3.71 vs 1.84 months, P=0.023). Median survival was 8.61 vs 5.29 months (P=0.209). [88] | [88] |
| HER2-Negative Breast Cancer | Neoadjuvant Chemotherapy (NAC) | nab-Paclitaxel vs. Docetaxel (both with epirubicin/cyclophosphamide) | nab-Paclitaxel achieved a significantly higher pathological complete response (pCR) rate (36.71% vs 20.00%, P=0.031), particularly in triple-negative and node-negative subtypes. No significant difference in disease-free survival was observed. [89] | [89] |
| Non-Small Cell Lung Cancer (NSCLC) | Second-line therapy | Paclitaxel poliglumex (PPX) vs. Docetaxel | Both agents showed equivalent median survival (6.9 months). PPX had less febrile neutropenia and alopecia, but increased neurotoxicity. [90] | [90] |
Table 3: Comparative Safety Profiles of Select Microtubule-Targeting Agents
| Adverse Event | Docetaxel | nab-Paclitaxel | Paclitaxel Poliglumex (PPX) | Weekly Paclitaxel (wPTX) |
|---|---|---|---|---|
| Grade 3/4 Neutropenia | High (58.3%) [88] | Not specified | Significantly less than docetaxel (P<0.001) [90] | Lower than docetaxel (28.6%) [88] |
| Febrile Neutropenia | 33.3% [88] | Not specified | Less than docetaxel (P=0.006) [90] | 7.1% [88] |
| Grade 3/4 Neuropathy | Less common [90] | Higher incidence (60.76% vs 36.25%) [89] | More common than docetaxel (P<0.001) [90] | Not specified |
| Arthralgia | More frequent (57.50%) [89] | Lower incidence (39.97%) [89] | Not specified | Not specified |
| Alopecia | Common [90] | Not specified | Less than docetaxel [90] | Not specified |
Experimental Protocols for Evaluating MTA Efficacy and Mechanism
Protocol 1: In Vitro Assessment of Cell Proliferation and Viability
Objective: To determine the anti-proliferative effects and IC₅₀ values of MTAs on cancer cell lines. Materials:
- Cell Lines: HER2-overexpressing breast cancer lines (e.g., BT-474, SK-BR-3) [91] [92] or triple-negative breast cancer lines (e.g., MDA-MB-231) [93].
- Test Agents: Stock solutions of MTAs (e.g., paclitaxel, vinblastine) and/or targeted inhibitors (e.g., lapatinib). Aliquots are stored at -20°C [92].
- Reagent: Cell proliferation reagent WST-1 [92].
Methodology:
- Cell Seeding: Seed cells into 96-well plates at a density of 4,000 cells per well in 90 μL of culture medium and incubate overnight [92].
- Drug Treatment: Add 10 μL of serially diluted MTA solutions to the wells to achieve the desired final concentration range (e.g., 0.001 to 1 μmol/L for lapatinib) [92].
- Incubation: Treat cells for a predetermined period (e.g., 72 hours) [92].
- Viability Assay: Add 10 μL of WST-1 reagent directly to each well. Incubate the plates at 37°C for 30 minutes to 4 hours [92].
- Absorbance Measurement: Measure the absorbance of the samples using a plate reader at 450 nm. The signal correlates with the number of metabolically active cells [92].
- Data Analysis: Calculate cell viability as a percentage of the untreated control. Determine the half-maximal inhibitory concentration (IC₅₀) using non-linear regression analysis of the dose-response curve [92].
Protocol 2: Analysis of Mitotic Defects via Live-Cell Imaging
Objective: To dynamically assess the impact of MTAs on mitotic progression and induction of mitotic arrest. Materials:
- Cell Lines: Breast cancer cell lines (e.g., MDA-MB-231, CAL-51, T-47D) [93].
- Inhibitors: MTAs (e.g., paclitaxel, vinblastine) and/or pathway inhibitors (e.g., CMPD1, MK2-IN-3) [93].
- Equipment: Microscope with live-cell imaging capabilities and an environmental chamber to maintain 37°C and 5% CO₂ [93].
Methodology:
- Cell Preparation: Seed cells into multi-well imaging plates and allow them to adhere.
- Treatment and Imaging: Treat cells with the MTA at a clinically relevant or subclinical concentration (e.g., 10 nM CMPD1 or 5-50 nM paclitaxel) and immediately place the plate under the microscope [93].
- Image Acquisition: Capture images at high temporal resolution (e.g., every 5-10 minutes) for 24-48 hours to track cell division.
- Phenotypic Scoring: Analyze the images for key mitotic events:
Protocol 3: Evaluating Efficacy in Combination Therapies
Objective: To investigate synergistic effects between MTAs and targeted signaling pathway inhibitors. Materials:
- Test Agents: Microtubule destabilizer (e.g., Vinblastine) and a specific p38-MK2 pathway inhibitor (e.g., MK2-IN-3) [93].
- Assay: Phosphorylation array assays and Western blot analysis for downstream pathway analysis [91].
Methodology:
- Cell Treatment: Pre-treat cancer cells with a subclinical concentration of an MK2 inhibitor [93].
- Combination Treatment: Co-treat cells with the MK2 inhibitor and a subclinical concentration of a microtubule destabilizer like vinblastine [93].
- Outcome Assessment:
- Phenotypic Analysis: Use live-cell imaging (as in Protocol 2) to quantify the enhancement of mitotic defects [93].
- Mechanistic Analysis: Perform Western blot analysis to confirm inhibition of the target pathway (e.g., reduced phosphorylation of MK2 and its substrates) and modulation of proteins like ERα and phosphorylated Akt [91].
Signaling Pathways and Novel Therapeutic Combinations
The efficacy of MTAs can be significantly modulated by interacting signaling pathways within the cell. Combining MTAs with targeted inhibitors of these pathways represents a promising strategy to overcome resistance and enhance antitumor activity.
The p38-MK2 Pathway as a Modulator of MTA Efficacy
Research has shown that inhibition of the p38 MAPK-MK2 signaling pathway can sensitize cancer cells to MTA treatment. The small molecule CMPD1, initially identified as an MK2 inhibitor, was found to also induce microtubule depolymerization, leading to irreversible mitotic defects in cancer cells at low concentrations (10 nM) [93]. Furthermore, combining a specific MK2 inhibitor (MK2-IN-3) with vinblastine, a microtubule destabilizer, resulted in a significantly increased efficacy in inducing mitotic defects compared to either agent alone [93]. This suggests that the p38-MK2 pathway promotes survival in response to MTA-induced stress, and its inhibition is a potent synergistic strategy.
Diagram 2: Synergistic effect of combining microtubule-targeting agents with MK2 pathway inhibitors.
Targeting Receptor Tyrosine Kinases in Combination with MTAs
In HER2-positive breast cancers, the interplay between receptor tyrosine kinase signaling and the cytoskeleton is critical. Lapatinib, a dual tyrosine kinase inhibitor of EGFR and HER2, has shown combinatorial potential. Preclinical studies demonstrate that lapatinib inhibits downstream signaling pathways like MAPK and PI3K-AKT, increases pro-apoptotic proteins, and can restore sensitivity to antiestrogens [91] [94]. Furthermore, the calcitriol analog EB1089 can enhance the antiproliferative response of lapatinib combined with antiestrogens in HER2-positive breast cancer cells by modulating ERα expression and suppressing Akt phosphorylation [91]. Interestingly, the activity of lapatinib in HER2-overexpressing cells is primarily mediated through HER2 inhibition, independent of EGFR expression levels, as shown by siRNA knockdown experiments [92].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for MTA and Cytoskeletal Research
| Reagent / Assay | Function/Application | Example Usage in MTA Research |
|---|---|---|
| WST-1 Cell Proliferation Assay | Measures cellular metabolic activity as a surrogate for cell viability and proliferation. | Determining IC₅₀ values for MTAs like lapatinib in breast cancer cell lines [92]. |
| siRNA SMART pools | Enables targeted knockdown of specific genes to study their functional role. | Validating that lapatinib's activity is dependent on HER2, not EGFR, in HER2+ breast cancer cells [92]. |
| Phospho-Specific Antibodies for Western Blot | Detects activation/phosphorylation states of signaling proteins. | Confirming inhibition of AKT and ERK1/2 phosphorylation downstream of EGFR/HER2 after lapatinib treatment [91] [92]. |
| Live-Cell Imaging Systems | Allows real-time, dynamic observation of cellular processes like mitosis. | Quantifying the duration of prometaphase arrest induced by compounds like CMPD1 or paclitaxel [93]. |
| p38-MK2 Pathway Inhibitors | Chemically inhibits the p38-MK2 signaling axis to study its function. | Demonstrating synergistic enhancement of MTA efficacy (e.g., with vinblastine) [93]. |
The cytoskeleton serves as a critical determinant of cellular architecture and behavior, with its reorganization being a hallmark of invasive cancer cells. Plectin, a versatile cytoskeletal crosslinker protein, has emerged as a prominent target in mechanistic studies of invasion and metastasis. Research has demonstrated that plectin levels are significantly elevated in liver tumors, where its expression correlates with poor prognosis for hepatocellular carcinoma (HCC) patients [13]. Genetic and pharmacological inactivation of plectin potently suppresses the initiation, growth, and metastatic spread of HCC, establishing it as a validated therapeutic target [13]. This guide provides a comprehensive comparison of CRISPR/Cas9 technologies and knockout models for target identification and validation, with a specific focus on cytoskeletal proteins like plectin within the context of invasive cancer research.
Comparative Analysis of CRISPR/Cas9 Knockout Approaches
Performance Metrics of Optimized Gene Knockout Systems
Table 1: Comparison of CRISPR/Cas9 Knockout Efficiency Across Methodologies
| System Type | Single-Gene KO Efficiency (% INDELs) | Double-Gene KO Efficiency | Large Fragment Deletion Efficiency | Key Advantages | Notable Limitations |
|---|---|---|---|---|---|
| Inducible Cas9 (iCas9) in hPSCs [95] | 82-93% | >80% for double-gene KO | Up to 37.5% homozygous deletion | Tunable nuclease expression; Cost-effective | Requires optimization of multiple parameters |
| Dual-targeting sgRNA Libraries [96] | N/A - Enhanced essential gene depletion | Stronger depletion of essential genes vs. single-targeting | N/A | More effective knockout than single guides; Enables library size compression | Potential heightened DNA damage response |
| Commercial Plectin KO Kit [97] | Varies with biological complexity | Not specified | Not specified | Includes validated gRNAs and donor DNA; Pre-designed reagents | Proprietary system; Higher cost |
Benchmarking sgRNA Design Algorithms
The selection of effective single-guide RNAs (sgRNAs) remains a critical factor in successful gene knockout. A systematic evaluation of sgRNA design algorithms revealed that Benchling provided the most accurate predictions among tested platforms [95]. However, independent benchmarking of genome-wide libraries has identified Vienna Bioactivity CRISPR (VBC) scores as superior predictors of sgRNA efficacy, with guides selected using these scores demonstrating stronger depletion of essential genes in lethality screens [96].
Notably, even with high INDEL efficiency (≥80%), certain sgRNAs may fail to eliminate target protein expression, designating them as "ineffective sgRNAs" [95]. This underscores the necessity of empirical validation through Western blotting or other functional assays, particularly when working with cytoskeletal targets like plectin where functional knockout is essential for phenotypic assessment.
Experimental Protocols for Target Validation
Optimized Protocol for hPSCs with Inducible Cas9
The following methodology was optimized for high-efficiency gene knockout in human pluripotent stem cells (hPSCs) and can be adapted for cytoskeletal target validation [95]:
Cell Line Generation:
- Create doxycycline-inducible spCas9-expressing hPSCs (hPSCs-iCas9) by inserting the doxycycline-spCas9-puromycin cassette into the AAVS1 (PPP1R12C) locus using co-electroporation of two vectors at a 1:1 weight ratio.
- Use program CA137 on 4D-Nucleofector (Lonza Bioscience) for electroporation.
- Select with 0.5 μg/ml puromycin for one week post-nucleofection, then subclone and genotype surviving clones by junction PCR and Western blot.
sgRNA Design and Synthesis:
- Design sgRNAs using the CCTop algorithm or Benchling platform.
- Utilize chemically synthesized and modified sgRNAs (CSM-sgRNA) with 2'-O-methyl-3'-thiophosphonoacetate modifications at both 5' and 3' ends to enhance intracellular stability.
- For multiple gene targeting, conduct nucleofection with two or three sgRNAs at the same weight ratio to a fixed total amount of 5 μg.
Nucleofection Procedure:
- Dissociate Dox-induced hPSCs-iCas9 with EDTA and pellet by centrifugation at 250 g for 5 minutes.
- Combine sgRNA or sgRNA/ssODN mix with nucleofection buffer (P3 Primary Cell 4D-Nucleofector X Kit).
- Electroporate cell pellets using program CA137 on Lonza Nucleofector.
- For enhanced efficiency, perform repeated nucleofection 3 days after the first procedure following the same protocol.
Validation and Ineffective sgRNA Detection:
- Analyze Sanger sequencing chromatograms using ICE (Inference of CRISPR Edits) or TIDE algorithms.
- Confirm knockout at the protein level by Western blotting to detect ineffective sgRNAs that generate INDELs but fail to eliminate protein expression.
Phenotypic Characterization of Cytoskeletal Knockouts
For cytoskeletal targets like plectin, comprehensive phenotypic validation is essential:
Functional Assays:
- Conduct migration and invasion assays using standardized scratch tests or Transwell systems [13].
- Evaluate anchorage-independent proliferation through soft agar assays.
- Assess mitochondrial dysfunction in knockout cells via Mito stress assay (Seahorse) for metabolic profiling [95].
Cytoskeletal Architecture Analysis:
- Employ immunofluorescence microscopy to visualize cytoskeletal reorganization.
- Utilize computational pipelines to quantify cytoskeletal features including fiber orientation, compactness, radiality, and morphology [1].
- Apply automated feature extraction to distinguish unique microtubule signatures associated with invasive potential [1].
Signaling Pathways in Cytoskeletal-Targeted Knockouts
Plectin-Dependent Mechanosignaling in HCC
Genetic ablation of plectin in hepatocellular carcinoma models disrupts critical oncogenic signaling pathways through its function as a cytoskeletal crosslinker [13]. The diagram below illustrates the mechanistic relationship between plectin knockout and attenuated tumor signaling.
Figure 1: Plectin Knockout Disrupts Oncogenic Signaling in HCC
Proteomic and phosphoproteomic profiling following plectin inactivation demonstrates attenuation of FAK, MAPK/Erk, and PI3K/Akt signaling signatures [13]. This multifaceted disruption of mechanosensitive pathways highlights how cytoskeletal-targeted knockouts can simultaneously impair multiple hallmarks of cancer, effectively suppressing tumor growth, invasion, and metastatic outgrowth.
Experimental Workflow for Genetic Validation
Integrated Pipeline for Target Identification and Validation
The following workflow outlines a systematic approach for genetic validation of cytoskeletal targets using CRISPR/Cas9 knockout models, integrating computational and experimental methods.
Figure 2: Genetic Validation Workflow for Cytoskeletal Targets
This integrated approach begins with target selection informed by cytoskeletal organization analysis in invasive versus non-invasive cells [1]. Subsequent steps leverage optimized sgRNA design algorithms [95] [96] and efficient knockout generation protocols, culminating in phenotypic characterization using computational cytoskeletal feature extraction [1] and functional assays.
Research Reagent Solutions for Cytoskeletal Target Validation
Table 2: Essential Research Reagents for Genetic Validation Studies
| Reagent / Resource | Function / Application | Key Features / Specifications | Source / Reference |
|---|---|---|---|
| hPSCs-iCas9 Cell Line | Inducible CRISPR/Cas9 platform | Doxycycline-inducible spCas9; AAVS1-safe harbor locus | [95] |
| CSM-sgRNA | Chemically modified sgRNA | 2'-O-methyl-3'-thiophosphonoacetate modifications; Enhanced stability | GenScript Corporation [95] |
| Plectin (PLEC) Human Gene Knockout Kit | Targeted plectin knockout | 2 gRNA vectors + linear donor; EF1a-GFP-P2A-Puro selection | OriGene (Cat# KN411981) [97] |
| Plecstatin-1 (PST) | Pharmacological plectin inhibitor | Ruthenium-based compound; Mimics genetic ablation | Preclinical development [13] |
| Computational Cytoskeleton Pipeline | Quantitative cytoskeletal analysis | Analyzes orientation, compactness, radiality, morphology | [1] |
| PTP-01 Imaging Agent | Cell surface plectin detection | 111In-labeled tetrameric peptide; CSP-targeting | Phase 0 clinical study [98] |
The systematic comparison of CRISPR/Cas9 methodologies presented herein demonstrates that optimized gene knockout systems achieve exceptional efficiency (82-93% INDEL rates for single genes and over 80% for double-gene knockouts) when properly implemented [95]. The integration of computational sgRNA selection tools, particularly those utilizing VBC scores [96], with empirical validation through Western blotting provides a robust framework for identifying ineffective sgRNAs that fail to eliminate target protein expression despite high INDEL rates [95].
For cytoskeletal targets like plectin, the convergence of genetic knockout models with computational analysis of cytoskeletal architecture [1] and pharmacological inhibition strategies [13] creates a powerful multidimensional validation platform. This integrated approach enables researchers to not only establish causal relationships between target genes and invasive phenotypes but also to bridge fundamental discovery with therapeutic development through parallel assessment of genetic and pharmacological intervention strategies.
The cytoskeleton, a dynamic network of intracellular filaments, is fundamental to cellular architecture, mechanotransduction, and motility. In cancer biology, cytoskeletal remodeling is intrinsically linked to tumor invasion, metastasis, and therapeutic resistance. This guide objectively compares the performance of cytoskeletal markers and associated methodologies in prognosticating patient survival across multiple cancer types. We synthesize experimental data and analytical frameworks that establish cytoskeletal genes and organization signatures as powerful predictors of clinical outcomes, providing researchers with a comparative analysis of tools and biomarkers for dissecting the role of the cytoskeleton in cancer progression.
Quantitative Prognostic Correlations of Cytoskeletal Markers
The expression levels of specific cytoskeletal genes and the physical properties of cytoskeletal architecture have been quantitatively linked to survival outcomes in diverse malignancies. The tables below summarize key prognostic correlations identified in recent studies.
Table 1: Prognostic Cytoskeletal Gene Signatures in Solid Tumors
| Cancer Type | Gene(s) | Expression Direction | Prognostic Correlation | Hazard Ratio/Statistical Significance | Study Model |
|---|---|---|---|---|---|
| Hepatocellular Carcinoma (HCC) | 5-gene model (ARPC1A, CCNB2, CKAP5, DCTN2, TTK) | High | Poor Overall Survival | Validated in TCGA, ICGC LIRI-JP, and CHCC-HBV cohorts [99] | LASSO-Random Forest Prognostic Model |
| IDH Wild-type Glioma | FN1, HIF3A, EIF4B | High | Poorer Survival | HR: 1.40, 1.49, 1.54 (p < 0.05) [100] | Accelerated Failure Time (AFT) Model |
| IDH Wild-type Glioma | PTK2, CCND2, RAD51L3-RFFL, MAX | High | Protective Effect | HR: 0.76, 0.78, 0.79, 0.79 (p < 0.05) [100] | Accelerated Failure Time (AFT) Model |
| Breast Cancer (ER+/HER2-) | Activity-regulated cytoskeleton-associated protein (ARC) | High | Improved Survival | Better Disease-specific & Overall Survival (p < 0.05) [101] | Bulk RNA-seq Cohort Analysis |
Table 2: Quantitative Cytoskeletal Architecture Features in Cancer Cells
| Cellular Phenotype | Cytoskeletal Feature | Measurement | Quantitative Value | Prognostic/Functional Implication | Analysis Method |
|---|---|---|---|---|---|
| Mutant E-cadherin (Invasive) | Microtubule Orientation | Orientational Order Parameter (OOP) | Significantly Lower OOP [1] | Disorganized fibers, associated with invasive capacity [1] | Immunofluorescence & Computational Pipeline |
| Mutant E-cadherin (Invasive) | Microtubule Morphology | Fiber Length | Shorter fibers [1] | Disseminating cellular properties [1] | Immunofluorescence & Computational Pipeline |
| Mutant E-cadherin (Invasive) | Fiber Distribution | Compactness (Nl/Ac) | More compactly distributed [1] | Altered cellular mechanics [1] | Immunofluorescence & Computational Pipeline |
Experimental Protocols for Cytoskeletal Analysis
Computational Pipeline for Cytoskeletal Architecture Dissection
A novel image-based pipeline was developed to characterize the cytoskeletal architecture of cancer cells with invasive potential [1].
- Sample Preparation and Imaging: Cells are grown on laminin-coated surfaces to model cell-ECM interaction, fixed, and immunostained for cytoskeletal components (e.g., α-tubulin for microtubules) alongside nuclear staining. Multiple images are acquired for each channel along the Z-axis [1].
- Image Preprocessing: Z-stacks are projected into 2D using maximum intensity projection (MIP). Deconvolution is applied to remove noise and blur, improving contrast and resolution. A Gaussian filter smooths the fluorescence signal, a Sato filter highlights curvilinear structures, and a Hessian filter generates binary images [1].
- Feature Extraction: Binary images are skeletonized. The pipeline then automatically extracts two classes of features:
- Line Segment Features (LSFs): Quantify fiber morphology, including orientation (Orientational Order Parameter - OOP), length, quantity, compactness (number of fibers per cell area), and radiality relative to the nucleus centroid [1].
- Cytoskeleton Network Features (CNFs): Analyze higher-order topology, including bundling, parallelism, connectivity, and complexity, using graph networks where nodes represent fiber intersections [1].
- Validation: This framework was validated using cells expressing wild-type versus a deleterious mutant E-cadherin (p.L13_L15del), a known driver of invasive phenotypes, successfully distinguishing their unique microtubule signatures [1].
Network-Based Identification of Survival-Associated Genes
A network-based survival (netSurvival) method was used to identify prognostic gene expression markers in recurrent IDH wild-type gliomas [100].
- Data Preprocessing: Gene expression data (e.g., TPM values) from cohorts like the GLASS consortium are normalized. Genes with excessive zero expression are removed. Expression levels are dichotomized into high and low groups based on median values [100].
- Network Construction: For each gene, one-dimensional hierarchical clustering groups samples based on normalized expression. Patient clusters with the top 10% highest absolute expression form nodes in a gene-gene interaction network. Edges connect nodes that share patients, creating a network focused on gene-level interactions [100].
- Random Walk and Significance Testing: A random walk algorithm identifies paths of connected nodes where the associated patient groups show significantly different survival outcomes (assessed by log-rank test). Fisher's exact test is then used to identify genes whose presence is independent of these significant survival-associated paths, pinpointing prognostic markers [100].
- Validation: Identified markers are validated using regularized Cox models (Ridge, Lasso, Elastic Net) and by assessing the survival differences of stratified patient groups via prognostic indexes [100].
Machine Learning Framework for Cytoskeletal Gene Screening
An integrative machine learning and differential expression analysis workflow identifies cytoskeletal genes associated with age-related diseases and cancer [102] [99].
- Data Acquisition and Gene Set Definition: Transcriptomic data (e.g., from TCGA) and clinical information for the disease of interest are collected. A definitive list of cytoskeleton-related genes is retrieved from the Gene Ontology browser (GO:0005856) or the MSigDB database [102] [99].
- Model Construction and Feature Selection: Multiple machine learning algorithms (e.g., Support Vector Machines (SVM), Random Forest) are trained to classify patient and normal samples based on cytoskeletal gene expression. Recursive Feature Elimination (RFE) paired with the best-performing classifier (often SVM) is used to select the most discriminative subset of genes [102]. Alternatively, LASSO Cox regression is employed for prognostic model building in survival analysis [99] [103].
- Differential Expression and Integration: Differential expression analysis (e.g., using Limma or DESeq2) is conducted between patient and control groups. The final candidate genes are identified by focusing on the overlap between the machine-learning-selected features and the differentially expressed genes [102].
- Validation: The prognostic power of the identified gene signatures is validated using receiver operating characteristic (ROC) analysis on external datasets and survival analysis (Kaplan-Meier curves) in independent cohorts [102] [99].
Cytoskeletal Signaling Pathways in Cancer Prognosis
Cytoskeletal dynamics are regulated by key oncogenic signaling pathways. Their dysregulation contributes to invasive potential and poor survival, making them a focus for prognostic marker discovery and therapeutic targeting.
Diagram 1: Cytoskeleton-associated signaling pathways influencing cancer progression and patient survival. Key regulators (yellow) impact specific cytoskeletal processes (green) that collectively drive malignant phenotypes (red) linked to poor clinical outcomes [22] [99].
The Scientist's Toolkit: Research Reagent Solutions
This section details essential materials and tools used in the featured experiments for researchers seeking to implement these protocols.
Table 3: Key Research Reagents and Computational Tools for Cytoskeletal Prognostic Studies
| Item Name | Function/Application | Specific Use Case | Example/Reference |
|---|---|---|---|
| Anti-α-Tubulin Antibody | Immunofluorescence staining of microtubules | Visualizing and quantifying microtubule architecture in fixed cells [1] | Standard immunofluorescence protocol |
| Laminin-Coated Surfaces | Mimics extracellular matrix (ECM) for cell growth | Providing a physiologically relevant environment for studying cell-ECM interaction and cytoskeletal organization [1] | Cell culture substrate |
| Plasma Care Device (NIPP) | Generates non-invasive physical plasma | Inducing oxidative stress to disrupt tumor cell cytoskeleton and metabolism as an oncological therapy [104] | Terraplasma Medical device |
| GLASS Consortium Data | Longitudinal genomic data for gliomas | Studying gene expression patterns associated with recurrence and survival in IDH wild-type gliomas [100] | Publicly available dataset |
| TCGA/ICGC Datasets | Multi-omics and clinical data for various cancers | Training and validating machine learning models for prognostic cytoskeletal gene signatures [101] [102] [99] | Publicly available datasets (e.g., cBioPortal) |
| R/ Python (limma, DESeq2, glmnet, sklearn) | Statistical computing and machine learning | Performing differential expression analysis, feature selection, and building prognostic classification models [100] [102] [99] | Open-source software packages |
| Computational Pipeline (Custom) | Quantifying cytoskeletal features from images | Extracting metrics like OOP, compactness, and radiality from immunofluorescence images [1] | Custom scripts (Sato filter, Hessian filter, skeletonization) |
The emerging field of mechanomedicine represents a paradigm shift in therapeutic development, focusing on the fundamental role that mechanical forces play in health and disease. Mechanotransduction—the process by which cells convert mechanical signals into biochemical responses—regulates essential cellular functions including growth, differentiation, migration, and gene expression [105]. This process operates through an interconnected network of intracellular components, from cell-surface mechanosensitive ion channels and adhesion receptors to the cytoskeleton and nuclear machinery. Disruptions in mechanotransduction pathways contribute significantly to disease progression in conditions ranging from cancer and fibrosis to cardiovascular disorders and neurodegenerative diseases [105]. This review compares current therapeutic strategies targeting cytoskeletal dynamics and mechanotransduction, examining both molecular mechanisms and experimental approaches that are reshaping drug development for researchers and scientists in the field.
The therapeutic landscape is diversifying from traditional cytoskeletal inhibition toward more precise disruption of mechanotransduction signaling hubs. Cytoskeletal-targeting agents traditionally focus on direct disruption of actin or microtubule dynamics, while emerging mechanotherapeutic approaches aim for greater specificity by targeting mechanosensitive ion channels, adhesion complexes, or nuclear shuttling mechanisms [105]. This evolution reflects our growing understanding of how mechanical forces influence disease processes at cellular and tissue levels, opening new avenues for intervention that complement conventional biochemical targeting strategies.
Comparative Analysis of Cytoskeletal and Mechanotransduction Targets
Table 1: Comparison of Cytoskeletal and Mechanotransduction Targets
| Target Category | Specific Target | Therapeutic Approach | Experimental Evidence | Key Findings |
|---|---|---|---|---|
| Actin Cytoskeleton | Actin-tropomyosin binding | Small molecule inhibition [105] | Cell culture experiments [105] | Reduced tumor cell invasiveness |
| Actin stabilization | Blocking cofilin phosphorylation [105] | Cell culture models [105] | Potential application for Alzheimer's disease | |
| Signaling Kinases | ROCK1/ROCK2 | Inhibition with AT13148 [105] | Cell culture & mouse models [105] | Reduced cancer progression |
| Rho/ROCK pathway | Fasudil inhibition [105] | Clinical studies (pulmonary hypertension) [105] | Improved vascular function | |
| Mechanosensitive Transcription | YAP/TAZ-TEAD interaction | Disruption with VGLL4 or verteporfin [105] | Mouse & rat models [105] | Suppressed tumor growth & fibrosis |
| YAP hyperactivation | Genetic activation [105] | Mouse models [105] | Promoted organ regeneration | |
| Adhesion Complexes | Integrin αvβ3 | Small molecule antagonists [105] | Preclinical models [105] | Reduced tumor spread |
| Integrin αvβ6 | Antibody blockade [105] | Mouse models [105] | Attenuated fibrosis progression | |
| Nuclear Mechanotransduction | SUN1/SUN2 | SUN1 removal or SUN2 suppression [105] | Knockout mouse & cancer models [105] | Destabilized cell junctions, reduced cancer progression |
Table 2: Experimental Models for Mechanotherapeutic Development
| Experimental System | Key Measurable Parameters | Applications | Technical Advantages | Limitations |
|---|---|---|---|---|
| Photo-responsive hydrogels | Migration speed, traction forces, focal adhesion dynamics [106] | Cell migration on soft substrates [106] | Reversible rigidity switching, precise temporal control [106] | Simplified compared to native ECM |
| Molecular clutch models | Loading rate response, reinforcement kinetics, cytoskeletal softening [107] | Quantifying mechanosensing principles [107] | Predicts cellular response to force dynamics [107] | Computational abstraction of complex biology |
| DUOX2 redox signaling models | H2O2 flash localization, cytoskeletal remodeling, wound closure [108] | Epithelial migration & barrier repair [108] | Real-time visualization of spatiotemporal signaling [108] | Cell-type specific mechanisms |
| Stretch-based systems | YAP localization, adhesion growth, cytoskeletal organization [107] | Cardiovascular & pulmonary mechanobiology [107] | Applicable to various cell types & conditions [107] | May oversimplify tissue-level complexity |
Experimental Protocols for Key Mechanobiology Studies
Protocol: Dynamic Substrate Rigidity and Cell Migration
This methodology examines how cells respond to time-varying mechanical cues, challenging the traditional view that cells migrate poorly on soft substrates [106].
- Cell Preparation: Culture human mesenchymal stem cells (hMSCs) using standard conditions. Serum-starve cells for 24 hours before experimentation to minimize confounding effects of serum-induced signaling.
- Substrate Fabrication: Prepare photo-responsive yellow protein (PYP) hydrogels with immobilized RGD peptides (5 mM concentration) to ensure constant ligand density during modulus modulation [106].
- Rigidity Cycling: Subject hydrogels to cyclic illumination (1 min on/off intervals) for 12 hours using a controlled light source. Maintain consistent light intensity throughout experiments.
- Data Acquisition: Record cell movements using high-magnification time-lapse microscopy. Acquire parallel multicellular observations to provide population-level context.
- Analysis: Quantify migration speed, directional persistence, and morphological parameters (cell area, circularity, aspect ratio) using automated tracking software. Compare to static controls (dark condition: ~2.2 kPa; continuous illumination: ~1.6 kPa) [106].
Protocol: Force Loading Rate and Mechanosensing
This approach identifies the rate of force application (loading rate) as a key driver of mechanosensing, using both substrate stretching and molecular perturbation [107].
- Cell Seeding: Plate mouse embryonic fibroblasts on soft (0.6 kPa) fibronectin-coated polyacrylamide gels to establish baseline low-adhesion conditions.
- Stretch Application: Apply cyclic biaxial stretch using a cell-stretching device. Implement triangular waveform signals at varying frequencies (0.125-2 Hz) and amplitudes (2.5-20%) for 1 hour.
- Pharmacological Intervention: For cytoskeletal stabilization studies, treat cells with 1 μM Jasplakinolide for 2 hours prior to and during stretch application [107].
- Genetic Manipulation: Perform talin knock-down using RNA interference to confirm talin-dependent mechanosensing [107].
- Assessment: Fix cells and immunostain for YAP localization and paxillin-containing adhesions. Quantify nuclear-to-cytoplasmic YAP ratio and adhesion size distribution. Perform actin anisotropy analysis to quantify cytoskeletal organization [107].
Protocol: Redox Signaling and Cytoskeletal Dynamics
This procedure investigates how spatiotemporal H2O2 generation coordinates actin remodeling through DUOX2 localization and activity [108].
- Cell Model Establishment: Lentivirally transduce NCI-H661 cells (which lack endogenous DUOX1/2) with DUOXA2 plus either wild-type DUOX2 or catalytically inactive DUOX2 E843Q mutant [108].
- Live-Cell Imaging: Transfert cells with membrane-targeted H2O2 sensor (HyPer7-MEM) and perform time-lapse imaging to visualize H2O2 generation at sites of actin remodeling.
- TNT Induction: Serum-starve cells for 24 hours to trigger tunneling nanotube (TNT) formation. Treat with NOX/DUOX inhibitor GKT137831 (10 μM) as a negative control.
- Immunofluorescence Analysis: Fix cells and co-stain for DUOX2 with TNT markers (RAB11, Myo10, GAP43) and actin regulatory proteins (cortactin).
- Wound Healing Assay: Create linear scratches in confluent monolayers and monitor closure kinetics. Apply PIEZO1 agonists/antagonists to test mechanosensory input to DUOX2 activation [108].
Signaling Pathways in Mechanotransduction
Diagram 1: Integrated mechanotransduction and redox signaling pathways. Key mechanosensing pathways (yellow) convert extracellular forces into biochemical signals through integrins and focal adhesions. These regulate transcriptional effectors (red) including YAP/TAZ and MRTF/SRF. Parallel redox signaling (green) activates DUOX2 in response to mechanical cues, generating H₂O₂ that promotes actin remodeling through FER kinase and cortactin.
Diagram 2: Dynamic substrate-induced migration mechanism. Rapid cyclic changes in substrate rigidity trigger a cascade of mechanical events that bypass conventional mesenchymal migration requirements. This alternative migratory mode enables cells to overcome the limitations of soft environments where traditional traction forces are insufficient [106].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Mechanobiology Investigations
| Reagent/Category | Specific Examples | Research Application | Mechanistic Function |
|---|---|---|---|
| Photo-responsive Hydrogels | PYP-based hydrogels [106] | Dynamic substrate rigidity studies | Reversibly switchable stiffness (1.6-2.2 kPa) via light illumination |
| Mechanosensitive Ion Channel Modulators | PIEZO1 activators/inhibitors [108] | Calcium-mediated mechanosensing | Initiate mechanosensory signals that activate downstream effectors |
| ROCK Pathway Inhibitors | Fasudil, Y-27632, AT13148 [105] [107] | Cytoskeletal tension modulation | Inhibit Rho-associated kinase to reduce actomyosin contractility |
| YAP/TAZ Inhibitors | Verteporfin, VGLL4 expression [105] | Transcriptional mechanosignaling | Disrupt YAP/TAZ-TEAD interactions and nuclear localization |
| Genetically Encoded Biosensors | HyPer7-MEM, UnaG-DUOX2 [108] | Spatiotemporal redox signaling | Visualize H2O2 generation and DUOX2 trafficking in live cells |
| Cytoskeletal Stabilizing Agents | Jasplakinolide [107] | Actin dynamics perturbation | Prevent actin depolymerization to test cytoskeletal softening hypotheses |
| Focal Adhesion Markers | Paxillin, vinculin antibodies [107] | Adhesion complex monitoring | Quantify adhesion growth and turnover in response to mechanical cues |
| NOX/DUOX Inhibitors | GKT137831 [108] | Redox signaling dissection | Block NADPH oxidase activity to establish DUOX2-specific effects |
The comparative analysis of cytoskeletal inhibition and mechanotransduction disruption reveals an evolving therapeutic landscape with distinct advantages for each approach. Traditional cytoskeletal inhibitors offer well-characterized mechanisms and immediate effects on cell structure and motility, but often lack specificity and produce significant off-target effects. In contrast, emerging mechanotherapeutics provide greater precision by targeting specific mechanosensing components, potentially yielding improved therapeutic indices but requiring more sophisticated delivery and monitoring strategies [105].
Future directions in mechanomedicine will likely focus on multi-scale targeting that integrates cytoskeletal manipulation with precise disruption of key mechanosignaling hubs. The development of dynamic biomaterials that mimic time-varying mechanical environments [106] provides new platforms for screening mechanotherapeutic candidates under more physiologically relevant conditions. Additionally, the intersection between redox signaling and mechanobiology [108] reveals previously underappreciated connections that could be leveraged for combination therapies. As our understanding of mechanotransduction networks deepens, the next generation of therapeutics will likely move beyond single-target inhibition toward systems-level interventions that reset pathological mechanical signaling states while preserving physiological force sensing essential for tissue homeostasis.
Conclusion
The comparative analysis of cytoskeletal organization unequivocally establishes it as a defining feature of invasive cancer cells, with quantifiable signatures—such as disorganized, shorter microtubules and reconfigured actin networks—serving as potent biomarkers. The integration of advanced computational pipelines with physiologically relevant 3D models provides an unprecedented ability to decode these architectural blueprints. Validation through genetic and pharmacological studies, particularly on targets like plectin, confirms the cytoskeleton's role as a master regulator of oncogenic signaling and a viable therapeutic frontier. Future research must focus on translating these cytoskeletal signatures into non-invasive diagnostic tools and developing next-generation, targeted cytoskeletal drugs that maximize anti-metastatic efficacy while minimizing the off-target effects common to traditional cytotoxics. This integrative approach promises to open new avenues for combating metastasis, the primary cause of cancer mortality.
References
- 1. A novel computational approach to dissect the cytoskeletal ... [https://www.nature.com/articles/s41598-024-82538-w]
- 2. A new directionality tool for assessing microtubule pattern ... [https://pubmed.ncbi.nlm.nih.gov/24497496/]
- 3. Long-term potentiation-induced changes in actin dynamics ... [https://www.nature.com/articles/s42003-025-08459-0]
- 4. Regulation of actin cytoskeletal dynamics in T cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12185546/]
- 5. The tropomyosin 3.1/3.2 inhibitor ATM-3507 alters B-cell actin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12626929/]
- 6. Non-Invasive Physical Plasma as an Oncological Therapy ... [https://pubmed.ncbi.nlm.nih.gov/41228307/]
- 7. Impacts of structural properties of myosin II filaments on ... [https://elifesciences.org/articles/105236]
- 8. Force-activated zyxin assemblies coordinate actin ... [https://www.sciencedirect.com/science/article/pii/S0960982225000727]
- 9. Reconstituted systems for studying the architecture and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203972/]
- 10. The Role of Plectin Dysregulation in Cancer: Recent Advances [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472915/]
- 11. Plectin linkages are mechanosensitive and required for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9635290/]
- 12. Plectin-mediated cytoskeletal crosstalk as a target for ... [https://elifesciences.org/reviewed-preprints/102205v1/pdf]
- 13. Plectin-mediated cytoskeletal crosstalk as a target for ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11893104/]
- 14. Plectin-mediated cytoskeletal crosstalk as a target for ... [https://elifesciences.org/articles/102205]
- 15. Non-Invasive Physical Plasma as an Oncological Therapy ... [https://www.mdpi.com/2072-6694/17/21/3517]
- 16. The ECM and tissue architecture are major determinants of ... [https://www.nature.com/articles/s42003-023-05482-x]
- 17. E‐Cadherin Is a Structuring Component of Invadopodia in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077114/]
- 18. E-Cadherin: A Conductor of Cellular Signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12340774/]
- 19. Phases of tight junction barrier disruption during ... [https://www.nature.com/articles/s41598-025-96267-1]
- 20. Loss of E-Cadherin Alters Cigarette Smoke Extract (CSE) ... [https://pubmed.ncbi.nlm.nih.gov/41020432/]
- 21. Cytoskeleton-modulating nanomaterials and their ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X24001844]
- 22. Cytoskeletal dynamics in breast cancer: mechanistic ... [https://www.sciencedirect.com/science/article/pii/S2667005425001188]
- 23. Comparisons of actin filament disruptors and Rho kinase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3378243/]
- 24. Drug-Induced Changes of Cytoskeletal Structure and ... [https://www.sciencedirect.com/science/article/pii/S0006349500766148]
- 25. Disruption of the association between drug transporter and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5356828/]
- 26. Nellie: automated organelle segmentation, tracking and ... [https://www.nature.com/articles/s41592-025-02612-7]
- 27. Geometric feature enhanced line segment extraction from ... [https://www.sciencedirect.com/science/article/pii/S1569843222000607]
- 28. Genetically encoded reporters of actin filament ... [https://www.sciencedirect.com/science/article/abs/pii/S0092867425002764]
- 29. Metrics for Assessing Cytoskeletal Orientational Correlations ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004190]
- 30. Metrics for Assessing Cytoskeletal Orientational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4388480/]
- 31. Rapid quantification of mitochondrial fractal dimension in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6238904/]
- 32. The Quantitative Criteria Based on the Fractal Dimensions, ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2016.00034/full]
- 33. Quantitative phase microscopy for non-invasive live cell ... [https://www.nature.com/articles/s41598-021-83537-x]
- 34. Non-invasive kinetic modelling approaches for quantitative ... [https://link.springer.com/article/10.1007/s00259-022-06057-4]
- 35. Recent advances in 3D cell culture models in cancer drug ... [https://link.springer.com/article/10.1007/s40005-025-00740-y]
- 36. Bridging the gap: the role of 3D cell cultures in mimicking ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12378905/]
- 37. 2D versus 3D tumor-on-chip models to study the impact of ... [https://www.nature.com/articles/s41598-025-03504-8]
- 38. Breakthroughs and challenges of organoid models for ... [https://www.nature.com/articles/s41420-025-02505-w]
- 39. 3D Bioprinting and Artificial Intelligence for Tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12505267/]
- 40. Three-dimensional cell culture in vitro could better ... [https://www.sciencedirect.com/science/article/abs/pii/S0040816625005282]
- 41. Cracking the code: predicting tumor microenvironment ... [https://www.nature.com/articles/s44385-025-00032-y]
- 42. New NIH Project to Model Breast Tumors for Better ... [https://viterbischool.usc.edu/news/2025/10/new-nih-project-to-model-breast-tumors-for-better-immunotherapy/]
- 43. Challenges and opportunities for the next generation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12286354/]
- 44. Quantitative phase imaging with temporal kinetics predicts ... [https://www.nature.com/articles/s41467-025-61846-3]
- 45. Noninvasive real-time monitoring of cellular spatiotemporal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12266097/]
- 46. Label-free live cell recognition and tracking for biological ... [https://www.nature.com/articles/s44303-024-00046-y]
- 47. Label-Free, Single-Cell, Real-Time: Quantitative Phase ... [https://www.phasics.com/news/2025/08/22/label-free-single-cell-real-time-quantitative-phase-imaging-enables-functional-profiling-immune-cytotoxicity/]
- 48. Label-free differentiation of living versus dead single yeast ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00043b]
- 49. Optimization of Impedance-Based Real-Time Assay in ... [https://www.mdpi.com/2076-3417/15/15/8298]
- 50. Comparative Analysis of Dynamic Cell Viability, Migration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3477108/]
- 51. xCELLigence system for real-time label-free monitoring of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4061162/]
- 52. Comparison between xCELLigence biosensor technology and ... [https://josr-online.biomedcentral.com/articles/10.1186/s13018-017-0652-6]
- 53. Modulation of the Cytoskeleton for Cancer Therapy [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1681065/full]
- 54. Targeting and disrupting cytoskeleton using core-shell ... [https://www.sciencedirect.com/science/article/abs/pii/S0021979725026906]
- 55. The role of the cytoskeleton in cellular force generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3138199/]
- 56. Traction force analysis of cells cultured in 2D versus 3D ... [https://www.sciencedirect.com/science/article/pii/S2452199X25004323]
- 57. Matrix mechano-sensing at the invasive front induces a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11829002]
- 58. Impact of 2D versus 3D fibroblast models on Leishmania ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1654654/full]
- 59. A comparison of human skeletal muscle cell maturation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12166125/]
- 60. Comparison of 2D and 3D cell culture models [https://www.sciencedirect.com/science/article/abs/pii/S0003986124000390]
- 61. A Real-Time Cell Image Segmentation Method Based on Multi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383622/]
- 62. A self-supervised learning approach for high throughput ... [https://www.nature.com/articles/s42003-025-08190-w]
- 63. Automated smartphone based cell analysis platform [https://www.nature.com/articles/s44303-025-00093-z]
- 64. Advancing cell therapy manufacturing: an image-based ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1651144/full]
- 65. Advancing cell therapy manufacturing: an image-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12521239/]
- 66. Gene Expression Pattern Associated with Cytoskeletal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384099/]
- 67. Actin cytoskeleton dynamics affect replication of human ... [https://www.nature.com/articles/s41598-025-21385-9]
- 68. Cytoskeletal Drugs Modulate Off-Target Protein Folding ... [https://pubmed.ncbi.nlm.nih.gov/32567840/]
- 69. Dysregulation of Cytoskeleton Remodeling Drives Invasive ... [https://www.mdpi.com/2072-6694/13/22/5648]
- 70. Targeting the cytoskeleton as a therapeutic approach to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11034636/]
- 71. ROS-induced filament severing leads to surge in actin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11957145/]
- 72. Evaluating cell lines as tumour models by comparison of ... [https://www.nature.com/articles/ncomms3126]
- 73. Similarities and Differences in Gene Expression Networks ... [https://www.frontiersin.org/journals/artificial-intelligence/articles/10.3389/frai.2021.674370/full]
- 74. Cytoskeletal Organization Correlates to Motility and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7915464/]
- 75. Comprehensive comparison of molecular portraits between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5001206/]
- 76. Cancer cell lines evolve in ways that affect how they ... [https://www.broadinstitute.org/news/cancer-cell-lines-evolve-ways-affect-how-they-respond-drugs]
- 77. High-throughput single cell motility analysis using nanowell-in ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00478k]
- 78. ATLAS: Machine learning-enhanced filament analysis for ... [https://www.sciencedirect.com/science/article/pii/S2667074725000266]
- 79. Improving Methods for Diagnosing Esophageal Disorders [https://news.feinberg.northwestern.edu/2025/07/15/improving-methods-for-diagnosing-esophageal-disorders/]
- 80. Validation of stool and blood analysis compared to Inv A and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12228386/]
- 81. Correlating mechanical and gene expression data on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074148/]
- 82. The role of dynamic reciprocity in 3D cell migration [https://www.nature.com/articles/s44341-025-00027-1]
- 83. Inhibitors of cytoskeletal dynamics in malignant ... [https://www.oncotarget.com/article/27843/text/]
- 84. In vitro Cell Migration, Invasion, and Adhesion Assays [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2019.00107/full]
- 85. Actin cytoskeleton organization, cell surface modification ... [https://www.sciencedirect.com/science/article/abs/pii/S0014482714003413]
- 86. Microtubule-Targeting Agents: Advances in Tubulin Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347508/]
- 87. Recent Advances in Microtubule Targeting Agents for ... [https://www.mdpi.com/1420-3049/30/16/3314]
- 88. P-168: Retrospective comparison of efficacy and safety ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4983948/]
- 89. Efficacy and Safety of Albumin-Bound Paclitaxel Compared ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2021.760655/full]
- 90. Phase III trial comparing paclitaxel poliglumex vs docetaxel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2391114/]
- 91. EB1089 Increases the Antiproliferative Response of ... [https://pubmed.ncbi.nlm.nih.gov/38542136/]
- 92. Activity of lapatinib is independent of EGFR expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2525738/]
- 93. Inhibition of p38-MK2 pathway enhances the efficacy ... [https://elifesciences.org/articles/104859]
- 94. Efficacy and mechanism of action of the tyrosine kinase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4633889/]
- 95. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 96. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 97. Plectin (PLEC) Human Gene Knockout Kit (CRISPR) [https://www.origene.com/catalog/gene-expression/knockout-kits-crispr/kn411981-plectin-plec-human-gene-knockout-kit-crispr]
- 98. Imaging Cell Surface Plectin in PDAC Patients [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162372/]
- 99. Machine learning-assisted multi-dimensional ... [https://www.nature.com/articles/s41598-025-10056-4]
- 100. A network-based discovery of prognostic markers in ... [https://www.frontiersin.org/article/1672015]
- 101. Activity-Regulated Cytoskeleton-Associated Protein Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11750752/]
- 102. A computational framework for identifying cytoskeletal ... [https://www.nature.com/articles/s41598-025-97363-y]
- 103. Derivation of a novel multi‐gene prognostic model based on ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0328412]
- 104. Non-Invasive Physical Plasma as an Oncological Therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607350/]
- 105. Emerging mechanomedicines informed by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12158467/]
- 106. Dynamic rigidity changes enable rapid cell migration on soft ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12491448/]
- 107. The force loading rate drives cell mechanosensing through ... [https://www.nature.com/articles/s41467-021-24383-3]
- 108. Spatiotemporal H2O2 flashes coordinate actin cytoskeletal ... [https://www.nature.com/articles/s41467-025-62272-1]