A Step-by-Step Protocol for Microtubule Tactoids Self-Assembly: From Reconstitution to Analysis
This article provides a comprehensive guide for researchers and scientists on the self-assembly of microtubule tactoids, which are spindle-shaped, liquid crystal-like structures that serve as minimalistic in vitro models for...
A Step-by-Step Protocol for Microtubule Tactoids Self-Assembly: From Reconstitution to Analysis
Abstract
This article provides a comprehensive guide for researchers and scientists on the self-assembly of microtubule tactoids, which are spindle-shaped, liquid crystal-like structures that serve as minimalistic in vitro models for the mitotic spindle. It details a proven protocol utilizing the antiparallel crosslinker MAP65 and crowding agents to reconstitute these structures from a minimal set of components. The scope covers the foundational principles of tactoid formation, a step-by-step methodological pipeline, essential troubleshooting and optimization strategies based on recent research, and robust techniques for validating the assemblies through fluorescence microscopy and Fluorescence Recovery After Photobleaching (FRAP). This resource is tailored for professionals in biophysics and drug development seeking to implement this technique in their labs for studying cytoskeletal organization and screening potential therapeutic targets.
Understanding Microtubule Tactoids: Biological Significance and Liquid Crystal Principles
The mitotic spindle is a transient, complex machinery essential for cell division via mitosis and meiosis, responsible for the accurate segregation of genetic material [1] [2]. This structure is primarily composed of microtubule filaments—dynamic polymers of tubulin dimers with a high aspect ratio and significant stiffness—organized into specific architectures by microtubule-associated proteins (MAPs) and motor proteins [2].
The spindle contains distinct microtubule populations: kinetochore microtubules that connect spindle poles to chromosome kinetochores, and interpolar microtubules that grow past chromosomes and overlap at the spindle's central midzone [2]. A third type, astral microtubules, extends from the poles to the cell cortex and is outside the scope of this discussion [2]. The physical organization of short, dynamic, and overlapping microtubule arrays, particularly in meiotic spindles, gives these structures properties similar to liquid crystal tactoids [2]. These spindle-like shapes arise from the local alignment of asymmetric molecules or structures into a nematic phase, a property shared by the high local concentration of microtubules in the spindle [2].
Table 1: Key Features of the Mitotic Spindle
| Feature | Description | Biological Significance |
|---|---|---|
| Primary Function | Segregate chromosomes during cell division [2] | Ensures accurate genetic inheritance |
| Main Structural Element | Microtubules (tubulin polymers) [2] | Provides structural framework and force-generation machinery |
| Key Microtubule Types | Kinetochore, Interpolar, Astral [2] | Facilitates chromosome attachment, spindle stability, and positioning |
| Liquid Crystal Properties | Similar to a bipolar tactoid [2] | Suggests physical mechanisms contribute to self-organization |
The Rationale for In Vitro Models
Studying the mitotic spindle directly within living cells is complex due to the vast number of interconnected biochemical and physical variables. In vitro reconstitution provides a powerful alternative by simplifying the system to its core components, allowing for precise dissection of the mechanisms underlying spindle assembly and function [3].
A major goal of in vitro approaches is to understand how self-organization emerges from a minimal set of components. The cytoskeleton performs major internal reorganization without central direction, especially during mitosis [1] [2]. In vitro models enable researchers to test how specific proteins, such as antiparallel crosslinkers or molecular motors, contribute to the formation of bipolar structures from an initially disordered state [2] [4].
These models are also indispensable for functional analysis. For instance, the Xenopus laevis egg extract system allows for the direct inhibition of specific proteins via immunodepletion or dominant-negative constructs to analyze their mitotic function [3]. Similarly, isolated diatom spindles can be reactivated in vitro to study the energy requirements and mechanics of anaphase spindle elongation [5]. Ultimately, well-characterized in vitro systems serve as critical platforms for screening and drug discovery, enabling the identification of compounds that modulate spindle dynamics, which is relevant for cancer therapy [3].
Table 2: Comparison of Key In Vitro Models for Studying the Mitotic Spindle
| Model System | Key Components | Applications and Insights | Example Reference |
|---|---|---|---|
| Xenopus Egg Extracts | Meiotic cytoplasm, sperm nuclei or chromatin beads [3] [4] | Study of spindle assembly pathways, function via protein inhibition, and anaphase [3] | Sawin & Mitchison, 1991 [4] |
| Isolated Diatom Spindles | Pre-formed spindles isolated from cells [5] | Analysis of ATP-dependent spindle elongation and microtubule dynamics in anaphase B [5] | Cande & McDonald, 1985 [5] |
| Minimal Systems (e.g., Tactoids) | Purified tubulin, crosslinkers (MAP65), crowding agents [1] [2] | Investigation of physical principles of self-organization and minimal requirements for spindle-like shapes [2] | JoVE Protocol, 2022 [1] |
Pathways of Mitotic Spindle Assembly In Vitro
Research using in vitro models, particularly Xenopus egg extracts, has revealed multiple pathways for spindle assembly. One study identified two distinct pathways dependent on the cell cycle state of the extract when sperm nuclei are introduced [4].
In the first pathway, sperm nuclei added to metaphase-arrested extracts direct the assembly of polarized half-spindles. These half-spindles subsequently fuse pairwise to form bipolar spindles [4]. In the second pathway, sperm nuclei are introduced to extracts that are first induced to enter interphase and are then arrested in the subsequent mitosis. In this case, a single sperm nucleus can direct the assembly of a complete bipolar spindle [4]. A key finding from this work is that while microtubule arrays are strongly biased towards chromatin, the establishment of stable bipolar arrays does not strictly depend on specific kinetochore-microtubule interactions in either pathway [4]. This suggests a hierarchy of selective microtubule stabilization involving chromatin-microtubule and antiparallel microtubule-microtubule interactions [4].
The following diagram illustrates the logical relationship and key differences between these two assembly pathways.
The Microtubule Tactoid Model: A Minimal System
The microtubule tactoid model represents a minimalist approach to reconstituting spindle-like structures. This system aims to recreate the spindle's shape and some physical properties using a highly reduced set of components, focusing on the role of antiparallel microtubule crosslinking and macromolecular crowding [2].
The core insight behind this model is that microtubules, as mesoscale objects with high stiffness and a high aspect ratio, can be treated as scaled-up versions of liquid crystal molecules (mesogens) [2]. The meiotic spindle's behavior, including its ability to coalesce and merge, aligns with the properties of a liquid crystal tactoid—a nematic phase that nucleates and grows from an isotropic state [2]. The characteristic spindle shape arises because the asymmetric microtubules cannot form a rounded crystal when they align [2].
Successful formation of these tactoids requires specific conditions:
- Short Microtubules: Achieved by nucleating and stabilizing filaments with GMPCPP, a non-hydrolyzable GTP analog, to prevent dynamic instability and end-to-end annealing [2].
- Antiparallel Crosslinker: MAP65, a plant microtubule-associated protein and a homolog of mammalian PRC1 and yeast Ase1, is used to self-organize microtubules into spindle-like assemblies [1] [2] [6].
- Crowding Agent: Polyethylene glycol (PEG) is included to create depletion forces that increase the local concentration of microtubules and favor bundle formation [2].
A key advantage of this minimal system is that it is highly reproducible and accessible, requiring only purified components rather than complex biological extracts [7]. It serves as a bridge, connecting biological organization to the principles of soft matter physics [2] [7].
Research Reagent Solutions for Microtubule Tactoid Assembly
The following table details the essential reagents and their functions for recreating the microtubule tactoid assembly in vitro.
Table 3: Key Research Reagents for Microtubule Tactoid Assembly
| Reagent | Function and Purpose | Key Details |
|---|---|---|
| Tubulin | Core structural protein for microtubule polymerization [2] [7] | Often a mix of unlabeled and rhodamine-labeled tubulin for visualization [7] |
| MAP65 | Antiparallel microtubule crosslinker [1] [2] | Plant homolog of PRC1/Ase1; can be tagged with GFP for visualization [2] [7] |
| GMPCPP | Nucleates and stabilizes short microtubules [2] | A non-hydrolyzable GTP analog that caps microtubule ends [2] |
| PEG (Polyethylene Glycol) | Crowding agent [2] | Creates depletion interactions that promote microtubule condensation and bundling [2] |
| Glucose Oxidase/Catalase | Oxygen-scavenging system [7] | Prevents photodamage during fluorescence microscopy [7] |
| Pluronic F-127 | Non-ionic block copolymer surfactant [2] [7] | Coats chamber surfaces to prevent non-specific microtubule adhesion [2] [7] |
Detailed Protocol for Microtubule Tactoid Assembly and Analysis
This protocol is adapted from the JoVE video article "Self-Assembly of Microtubule Tactoids" [2] [7].
Tubulin Preparation
- Obtain an aliquot of unlabeled tubulin (1 mg lyophilized powder) from -80°C storage and keep it on ice.
- Resuspend the unlabeled tubulin in 200 µL of cold PEM-80 buffer. Keep on ice for 10 minutes to dissolve completely.
- Obtain a tube of rhodamine-labeled tubulin (20 µg lyophilized powder) from -80°C storage and keep it on ice.
- Resuspend the labeled tubulin in 4 µL of cold PEM-80 buffer. Keep on ice for 10 minutes.
- Combine 100 µL of the resuspended unlabeled tubulin with the 4 µL of resuspended rhodamine-labeled tubulin. Mix by pipetting slowly 6-7 times.
- Aliquot the tubulin mix into seven tubes (15 µL each), flash-freeze in liquid nitrogen, and store at -80°C for future use [7].
Flow Chamber Assembly
- Clean a glass slide thoroughly with double-distilled water and ethanol, then dry it with a lint-free wipe.
- Create two thin strips from double-sided tape and place them on the slide 5-8 mm apart.
- Place a silanized coverslip on top of the tape strips to create the flow path.
- Seal the chamber by gently pressing on the tape area with the back of a pen until the tape turns clear.
- Trim excess tape from the edges, leaving about 1 mm at the chamber entrance, and label the chamber [7].
Tactoid Assembly Reaction
- Thaw all reagents on ice and keep them on ice throughout the procedure.
- Coat the flow chamber with 20 µL of 5% Pluronic F-127 in PEM-80. Incubate in a humid chamber for 5-7 minutes.
- In a sterile tube on ice, mix the following components by pipetting 5-6 times:
- PEM-80 buffer
- GMPCPP
- Pluronic F-127
- Dithiothreitol (DTT)
- Glucose
- Polyethylene Glycol (PEG)
- The prepared tubulin mix
- MAP65 (with GFP-MAP65 for visualization)
- Add 1 µL of a pre-mixed glucose oxidase and catalase solution to the tubulin-MAP mixture. Mix by pipetting 7-8 times.
- Remove the Pluronic solution from the flow chamber by capillary action using a lint-free wipe at one end.
- Immediately add the tubulin-MAP mixture to the chamber entrance to draw it inside.
- Seal both ends of the chamber with five-minute epoxy.
- Incubate the chamber at 37°C for 30 minutes to nucleate and grow microtubule tactoids [7].
Imaging and Analysis
- Use a fluorescence microscope with a 60x or higher magnification objective and a numerical aperture (NA) of 1.2 or greater.
- Maintain the sample at 37°C during imaging using an environmental chamber or stage heater.
- For rhodamine-labeled tubulin, use a 561 nm laser with at least 1 mW of power at the sample.
- For GFP-MAP65, use a 488 nm laser for excitation.
- Acquire at least 10 images from different areas to capture over 100 tactoids. Save images as 16-bit TIFF files for analysis [7].
- To characterize constituent mobility, perform Fluorescence Recovery After Photobleaching (FRAP). For microtubules in tactoids, no fluorescence recovery is typically observed, indicating a solid-like state. For MAP65, fluorescence recovers gradually, indicating mobility, and can be fit to a rising exponential decay to find the amplitude and time scale of recovery [2] [7].
The experimental workflow for the entire protocol, from preparation to analysis, is summarized below.
What are Liquid Crystal Tactoids? Bridging Soft Matter Physics and Cell Biology
Liquid crystal tactoids are anisotropic microdroplets that spontaneously nucleate from an isotropic dispersion and represent a transition state before forming a macroscopic liquid crystalline phase [8] [9]. First observed in vanadium pentoxide sols by Zocher in 1925, these spindle-shaped or spherical birefringent microdroplets have since been identified in numerous lyotropic liquid crystalline systems including tobacco mosaic viruses, cellulose nanocrystals, carbon nanotubes, and various biological systems [8] [9]. Tactoids form through a process of microscopic phase separation when local concentration reaches a critical threshold, creating discrete ordered microdomains within a continuous disordered phase [8] [10]. What makes tactoids particularly fascinating to soft matter physicists and cell biologists alike is their ability to exhibit long-range orientational order while maintaining fluidity, a combination that enables complex structural transformations through coalescence, sedimentation, and response to external fields [8] [11].
The fundamental formation process of tactoids relies on a balance between competing forces: attractive interactions that gather mesogens into microdroplets and repulsive forces that arrange them into liquid crystalline order [8]. These interactions are often modeled using the Derjaguin-Landau-Verwey-Overbeek (DLVO) theory, where electrostatic repulsion between charged mesogens balances with long-range van der Waals attraction [8]. According to Onsager theory, the critical concentration for phase separation decreases with increasing aspect ratio of rod-like mesogens, meaning tactoids preferentially form in microdomains rich in high-aspect-ratio particles [8]. This selective nucleation process enables tactoids to function as molecular sorting mechanisms in both synthetic and biological systems.
Tactoids in Biological Systems
Microtubule Tactoids and Cell Division
The cytoskeleton exemplifies nature's utilization of liquid crystal principles, particularly during cell division when microtubules form the mitotic spindle without central direction [12]. This self-organizing process mirrors the behavior of liquid crystal tactoids, where microtubules function as mesoscale mesogens that assemble into spindle-like configurations [12] [1]. The meiotic spindle especially resembles a liquid crystal tactoid, containing short, dynamic microtubules that overlap in networked arrays [12]. This structural similarity has inspired researchers to reconstitute microtubule tactoids in vitro using minimal components, providing crucial insights into how cells might orchestrate such complex structural transformations without centralized control [12].
Microtubules possess inherent characteristics that make them ideal candidates for liquid crystalline organization: they are significantly longer than they are wide (high aspect ratio), exhibit substantial stiffness, and can align through steric and electrostatic interactions [12]. In the cellular environment, these properties enable microtubules to behave as scaled-up versions of liquid crystal molecules, following similar physical principles of phase transitions, nucleation, and growth [12]. The discovery that microtubules can form tactoids in vitro suggests that biological systems may exploit these fundamental soft matter principles to achieve complex organizational tasks.
Actin and Other Biological Tactoids
Beyond microtubules, tactoid formation has been observed in other crucial biological systems. Filamin has been shown to cause actin to condense into tactoids, suggesting similar liquid crystal principles may govern the organization of multiple cytoskeletal components [9]. Additionally, filamentous bacteriophage Pf4 generates a tactoid shell around host P. aeruginosa cells that confers antibiotic resistance [9]. These diverse biological examples demonstrate that tactoid formation is not merely a laboratory curiosity but a fundamental organizational strategy employed across biological systems.
Table 1: Biological Systems Exhibiting Tactoid Formation
| Biological System | Structural Role | Key Organizing Factors |
|---|---|---|
| Microtubule Spindles | Chromosome segregation during cell division | MAP65/Ase1/PRC1 crosslinkers, molecular crowding [12] |
| Actin Bundles | Cytoskeletal organization, mechanical stability | Filamin crosslinking, concentration thresholds [9] |
| Filamentous Phage Pf4 | Bacterial biofilm assembly, antibiotic resistance | Viral particle self-assembly at cell surface [9] |
Quantitative Characterization of Tactoids
Shape and Size Parameters
Tactoids exhibit predictable relationships between their size, shape, and internal structure. Experimental studies using β-lactoglobulin amyloid fibrils and cellulose nanocrystals have revealed that tactoids with smaller volumes (approximately 10² μm³) typically maintain a homogeneous configuration, while medium-sized tactoids (approximately 10³ μm³) often transition to a bipolar structure, and larger tactoids (approximately 10⁴ μm³) frequently develop cholesteric configurations with characteristic striped textures [11]. The aspect ratio (α = R/r, where R is the major semi-axis and r is the minor semi-axis) decreases with increasing volume, creating a predictable size-shape relationship [13].
The shape of tactoids is determined by the competition between surface energy, which favors spherical droplets, and elastic energy of the liquid crystalline phase, which promotes elongation [8]. For smaller tactoids, an additional competition emerges between surface energy and anchoring energy caused by deviation of the director field at the boundary from preferred orientation [8]. This delicate balance results in tactoids exhibiting spindle-like, prolate, or oblate shapes with different internal structures depending on their size and mesogen characteristics [8].
Relaxation Dynamics
The relaxation behavior of tactoids following deformation follows predictable exponential decay patterns. Research has demonstrated that shape relaxation, characterized by the parameter Ʀ = (R(t) - Requil)/(Rinit - Requil), follows a single exponential decay across all tactoid classes: Ʀ = exp(-t/τs) [11]. The characteristic shape relaxation time (τs) can be expressed as the sum of liquid crystalline anisotropic contributions (τa) and characteristic shape relaxation time of elongated isotropic tactoids (τ_i) [11]:
τs = b[ω/(ckBT(2K/K2 - 1)^(1/2)Mφ^(1/4)MQ^(3/4)ξ^(1/2)) + (βμI)/(bγ)]R_equiv
where K and K2 are elastic constants, Mφ and MQ are mobility parameters, ξ is the correlation length, μI is the isotropic viscosity, and γ is the interfacial tension [11].
Table 2: Characteristic Parameters of Different Tactoid Classes
| Tactoid Class | Typical Volume (μm³) | Aspect Ratio Range | Director Field Configuration | Relaxation Behavior |
|---|---|---|---|---|
| Homogeneous Nematic | ~10² | Higher | Uniform alignment parallel to long axis | Single exponential decay [11] |
| Bipolar Nematic | ~10³ | Intermediate | Director follows interface with defects at poles | Single exponential decay [11] |
| Uniaxial Cholesteric | ~10⁴ | Lower | Helical structure with periodic bands | Second-order exponential decay [11] |
Experimental Protocols
Microtubule Tactoid Self-Assembly Protocol
The following protocol enables the formation and study of microtubule tactoids in vitro using a minimal set of components, based on established methodologies [12] [1]:
Silanization of Cover Glasses
- Safety Note: Perform all steps in a fume hood while wearing appropriate protective equipment due to toxic silane vapors.
- Rinse cover glasses sequentially with ddH₂O, 70% ethanol, and ddH₂O to remove dust and soluble particles.
- Dry between rinses with lint-free laboratory wipes.
- Place cover glasses in a metal rack and expose to dimethyl-dichloro-silane vapor in a desiccator for 30 minutes.
- Bake silanized cover glasses at 110°C for 10 minutes to complete the process.
Preparation of Microtubules
- Use tubulin dimers purified from mammalian brain tissue or recombinant sources.
- Polymerize tubulin using GMPCPP (a non-hydrolyzable GTP analog) to nucleate and stabilize microtubules.
- Critical Step: Ensure microtubules are short (controlled length distribution) by regulating nucleation conditions and growth time.
- Stabilize resulting microtubules against dynamic instability and end-to-end annealing.
Formation of Tactoids
- Combine stabilized short microtubules with MAP65 (antiparallel microtubule crosslinker from plants) in appropriate buffer.
- Include PEG (crowding agent) to create depletion interactions that enhance local concentration.
- Incubate mixture between silanized cover glasses at room temperature for 30-60 minutes.
- Monitor tactoid formation using polarized light microscopy or fluorescence microscopy.
Characterization Methods
- Fluorescence Microscopy: Image tactoid shapes using fluorescently labeled microtubules.
- Fluorescence Recovery After Photobleaching (FRAP): Assess mobility of components within tactoids.
- Polarized Light Microscopy: Visualize birefringence patterns to determine internal director field.
Figure 1: Experimental workflow for microtubule tactoid assembly and characterization.
Capture and Stabilization of Tactoids in Solid Matrices
Studying the fine structure of tactoids has been challenging due to their highly deformable, fluid nature. Recent advances enable capturing these structures in solid matrices for detailed examination [8] [10]:
- Photopolymerization Method: Introduce cross-linked polyacrylamide matrices into the tactoid system.
- Rapid Polymerization: Initiate using UV light or chemical catalysts to quickly stabilize tactoid structures.
- Electron Microscopy: Examine captured tactoids using scanning electron microscopy (SEM) at resolutions sufficient to identify individual mesogens.
- Cross-section Analysis: Create fracture surfaces perpendicular to each other to examine chiral nematic structures from multiple angles.
This approach has revealed that small tactoids often exhibit unwound nematic structure with uniformly aligned cellulose nanocrystals, while larger tactoids develop left-handed helical chiral nematic structures appearing as series of flat periodic bands, each representing a half-helical pitch [8].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Microtubule Tactoid Research
| Reagent/Chemical | Function | Application Notes |
|---|---|---|
| MAP65 (Ase1/PRC1) | Antiparallel microtubule crosslinker | Plant-derived; essential for spindle-like assembly [12] |
| GMPCPP | Non-hydrolyzable GTP analog | Nucleates and stabilizes microtubules; controls length [12] |
| Polyethylene Glycol (PEG) | Crowding agent | Creates depletion interactions; enhances local concentration [12] |
| Dimethyl-dichloro-silane | Hydrophobic surface treatment | Enables polymer brush coating on cover glasses [12] |
| Tubulin Dimers | Structural protein | Purified from mammalian brain or recombinant sources [12] |
| β-lactoglobulin Amyloid Fibrils | Model rod-like colloids | Used for liquid crystalline phase studies [11] [13] |
| Cellulose Nanocrystals | Model rod-like colloids | Used for chiral nematic phase studies [8] [11] |
Advanced Applications and Research Implications
Flow-Induced Order-Order Transitions
Recent research has revealed that tactoids undergo fascinating structural transformations under flow conditions. When subjected to extensional flow in microfluidic devices, tactoids align and deform, undergoing order-order transitions at critical elongation thresholds [13]. Bipolar and cholesteric tactoids transform into homogeneous configurations when sufficiently extended, with the cholesteric pitch decreasing as an inverse power-law of the tactoid aspect ratio [13]. These flow-induced transformations provide insights into how biological systems might control structural organization through mechanical forces and confinement.
Microfluidic approaches with hyperbolic contraction zones enable precise control over extension rates (typically 0.004-0.020 s⁻¹ for amyloid fibril tactoids), allowing researchers to observe how tactoids respond to defined flow fields [13]. This methodology has revealed that tactoids align parallel to flow during extension, then rotate approximately 90° to perpendicular alignment after exiting extension zones, exhibiting behaviors similar to other anisotropic colloidal particles [13].
Nanoparticle Separation and Magnetic Manipulation
Liquid crystalline tactoids exhibit remarkable size-selective exclusion effects on foreign nanoparticles, providing a potential mechanism for nanoparticle separation [10]. Experiments with polymer nanospheres, gold nanoparticles, and paramagnetic nanoparticles have demonstrated that tactoids selectively incorporate particles below a threshold size while excluding larger particles [10]. This principle enables size-based separation where smaller nanoparticles are transported from disordered to ordered phases during phase separation.
When paramagnetic nanoparticles are added to systems containing tactoids, the disordered phases develop higher volume magnetic susceptibility due to preferential nanoparticle exclusion from tactoids [10]. This property enables control of tactoid movement and orientation using gradient magnetic fields as weak as several hundred Gauss/cm, potentially allowing researchers to direct phase separation rates, configurations, and director field orientations [10].
Figure 2: Tactoid applications in nanoparticle separation and magnetic control.
Liquid crystal tactoids represent a fascinating convergence of soft matter physics and biological organization principles. Their ability to self-assemble, transform, and respond to environmental cues makes them ideal model systems for understanding how biological systems achieve complex organization without central direction. The experimental protocols outlined here, particularly for microtubule tactoids, provide researchers with robust methodologies to explore these structures in controlled settings.
Future research directions will likely focus on harnessing tactoid properties for advanced materials design, drug delivery systems, and organizational principles in synthetic biology. The ability to control tactoid formation, transformation, and interaction with nanoparticles suggests numerous applications in biosensing, materials separation, and biomimetic material design. As our understanding of these remarkable structures deepens, we anticipate increasing convergence between fundamental soft matter research and biological applications, potentially revealing new principles of self-organization across length scales.
The cytoskeleton is a prime example of self-organization within the cell, achieving complex structural changes without central direction. This is particularly evident during cell division, where microtubules (MTs) form the mitotic spindle to segregate genetic material. Microtubule tactoids are spindle-shaped, self-organized assemblies that serve as minimal in vitro models for studying the physical principles underlying spindle formation [2] [12]. These structures recapitulate the bipolar morphology of the spindle midzone and are nucleated through a carefully balanced interplay of core biochemical components. The self-assembly process is driven by a minimal system comprising tubulin, antiparallel microtubule crosslinkers from the MAP65/PRC1/Ase1 family, and macromolecular crowding agents [2]. This application note details the protocols and quantitative data essential for reconstituting these structures, providing researchers with a toolkit for investigating cytoskeletal self-organization.
The Key Players: Components and Functions
Tubulin: The Structural Monomer
Tubulin, a heterodimeric protein, is the fundamental building block of microtubules. The kinetics of its polymerization are critical for tactoid formation.
- Self-Assembly Kinetics: Traditional one-dimensional (1D) models of microtubule growth assume a constant subunit dissociation rate. However, recent high-resolution studies using TIRF microscopy and laser tweezers have demonstrated that the tubulin subunit dissociation rate from a microtubule tip actually increases with free tubulin concentration [14]. This results in much faster on-off kinetics than previously estimated—an order of magnitude higher—and must be considered when modeling tactoid assembly.
- Microtubule Length Control: For successful tactoid formation, short microtubules are essential [2]. Long microtubules typically form extended bundles rather than finite, tapered tactoids. To obtain short, stable microtubules, the protocol recommends nucleating filaments with GMPCPP, a non-hydrolyzable GTP analog that suppresses dynamic instability and end-to-end annealing, resulting in a high density of short seeds for self-assembly [2].
MAP65/PRC1/Ase1: The Antiparallel Crosslinker
The MAP65/PRC1/Ase1 family of microtubule-associated proteins are the primary architects defining the architecture of microtubule tactoids.
- Function as Antiparallel Crosslinkers: These proteins are dimeric and specifically crosslink microtubules in an antiparallel orientation [2]. This activity is crucial for organizing microtubules into the bipolar, spindle-like shape of a tactoid, mirroring their role in forming the central spindle midzone during anaphase in cells [15].
- Liquid Condensate Formation: Recent findings indicate that MAP65 and its homolog PRC1 can undergo phase separation to form biomolecular condensates under physiological conditions, even without added crowding agents [16]. These condensates can nucleate and grow microtubule bundles. The size of the resulting microtubule asters is directly controlled by the concentration of MAP65, highlighting a potential mechanism for regulating intracellular organization [16].
- Regulatory Network: The activity of these crosslinkers is finely tuned by a post-translational modification network involving key kinases such as Cdk1, Plk1, and Cdc14, which ensures their function is restricted to the correct stage of cell division [15].
Crowding Agents: Creating the Physiochemical Environment
Macromolecular crowding agents are indispensable for recreating the dense cytoplasmic environment in vitro.
- Depletion Forces: Crowding agents like polyethylene glycol (PEG) create an "excluded volume" effect, which generates entropic (depletion) forces that promote microtubule bundling and condensation [17] [2]. This effect is critical for concentrating tubulin and MAP65 to the thresholds required for tactoid nucleation.
- Acceleration of Kinetics: Crowding dramatically accelerates biochemical reactions. In the case of microtubule growth, the presence of crowding agents can increase the apparent rate constant for tubulin addition by up to 10-fold [17]. When combined with regulatory enzymes like XMAP215 and EB1, crowding agents can push microtubule growth rates to approximately 45 μm/min at 10 μM tubulin, approaching the theoretical maximum and observed physiological rates [17].
Table 1: Key Components for Microtubule Tactoid Assembly
| Component | Key Function | Critical Considerations for Protocol Design |
|---|---|---|
| Tubulin | Structural monomer for microtubule polymerization | Use GMPCPP to generate short, stable seeds. Net growth rate increases with free tubulin concentration, but dissociation rate also rises [14]. |
| MAP65/PRC1/Ase1 | Antiparallel microtubule crosslinking | Concentration controls organization; low levels yield tactoids, high levels can form condensates that nucleate asters [2] [16]. |
| Crowding Agent (e.g., PEG) | Induces bundling via depletion forces & accelerates growth | Strength of attractive force and system density favor formation of extensile nematic networks over asters [17] [18]. |
Quantitative Data and Experimental Parameters
Successful self-assembly of microtubule tactoids depends on precise control over component concentrations and environmental conditions. The following tables consolidate key quantitative parameters from foundational studies.
Table 2: Effects of Crowding Agents and MAP65 on Microtubule Growth and Organization
| Experimental Condition | Observed Effect on Microtubules | Quantitative Measurement / Outcome |
|---|---|---|
| Crowding (PEG) + XMAP215 + EB1 | Synergistic acceleration of growth | Growth rates saturate near ~45 μm/min at 10 μM tubulin [17]. |
| Crowding Agent Alone | Increased tubulin addition rate | Apparent rate constant for tubulin addition increased up to 10-fold [17]. |
| MAP65 Crosslinking | Formation of finite, spindle-shaped tactoids | Requires short MTs; forms solid-like, birefringent, homogeneous tactoids [2]. |
| High [MAP65] / PRC1 | Formation of phase-separated condensates | Condensates nucleate MT bundles/asters; can be liquid-like (gelating over time) [16]. |
| Varying Ionic Strength | Modulation of MAP65-MT binding and organization | High salt shifts tactoids to unbounded bundles; reduces but does not eliminate MAP65 binding [16]. |
Table 3: Optimized Concentration Ranges for Tactoid Self-Assembly
| Parameter | Recommended Range / Value | Notes and Functional Impact |
|---|---|---|
| Tubulin Concentration | 0.5 - 2.0 μM (for growth) | Higher concentrations increase net growth but also dissociation fluctuations [14]. |
| MAP65 Concentration | Low, precisely titrated | Critical for tactoid shape; high concentrations promote large-scale aster formation [2] [16]. |
| Crowding Agent (PEG) | Variable, system-dependent | Strength of depletion attraction controls transition from asters to extensile bundles [18]. |
| Microtubule Length | Short (stabilized with GMPCPP) | Essential for tapered tactoids; long filaments form unbounded bundles [2]. |
| Ionic Strength | Physiological, but can be manipulated | Increased salt disrupts electrostatic interactions, affecting tactoid finiteness [16]. |
Detailed Experimental Protocol
This protocol outlines the steps for forming microtubule tactoids, with special emphasis on surface preparation to prevent non-specific adhesion and allow for proper microscopic observation.
Coverslip Silanization and Chamber Preparation
NOTE: Perform these steps in a fume hood while wearing appropriate personal protective equipment. Dimethyldichlorosilane (DDS) is highly toxic [2].
- Cleaning: Rinse coverslips sequentially with ddH₂O, 70% ethanol, and ddH₂O. Dry them with lint-free laboratory wipes between each rinse.
- UV-Ozone Cleaning: Place coverslips in a metal rack and transfer to a UV-Ozone (UVO) machine. Irradiate for 20 minutes to remove organic contaminants and reduce background fluorescence. (A plasma cleaner can be used as an alternative.)
- Solvent Rinsing: Using tweezers, transfer the coverslips to a dedicated silanization rack.
- Immerse the rack in 100% acetone for 1 hour. Rinse the container 3x with tap water and 3x with ddH₂O.
- Immerse the rack in 100% ethanol for 10 minutes. Rinse the container 3x with tap water and 3x with ddH₂O.
- Base Wash: Immerse the rack in 0.1 M KOH for 15 minutes. Rinse the container thoroughly 3x with tap water and 3x with ddH₂O.
- Final Water Rinse: Immerse the rack in ddH₂O three times for 5 minutes each.
- Drying: Air-dry the rack with coverslips overnight in a fume hood or laminar flow hood.
- Silanization: After complete drying, immerse the rack and coverslips in 2% dimethyldichlorosilane (DDS) in a dedicated container for 5 minutes.
- Ethanol Rinse: Immerse the rack and coverslips twice in 100% ethanol for 5 minutes each.
- Curing: Air-dry the coverslips again. The silanized coverslips are now ready for creating a flow chamber and subsequent polymer brush coating to create a non-adhesive passivated surface for imaging [2].
Tactoid Self-Assembly Reaction
- Prepare Tubulin Solution: Mix tubulin in a general tubulin buffer (e.g., 80 mM PIPES pH 6.8-7.0, 1 mM EGTA, 1-2 mM MgCl₂). For short, stable microtubules, include GMPCPP to nucleate and stabilize filaments against dynamic instability [2].
- Initiate Assembly: Combine the prepared tubulin solution with the antiparallel crosslinker MAP65 and a crowding agent (e.g., PEG) in the appropriate buffer.
- Incubate: Allow the reaction mixture to incubate at room temperature or a defined physiological temperature (e.g., 25-37°C) for a sufficient period (typically 30-60 minutes) for tactoids to nucleate and grow.
- Image: Transfer an aliquot of the reaction to a passivated imaging chamber and analyze using fluorescence microscopy. For characterization of shape and internal dynamics, employ:
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions and Materials
| Item / Reagent | Function / Role in Experiment | Example Source / Specification |
|---|---|---|
| Tubulin Protein (various species) | Core structural monomer for microtubule polymerization. | Porcine brain, human (e.g., MCF-7 cells), plant (soybean), fungi (Agaricus bisporus) [19]. |
| GMPCPP | Non-hydrolyzable GTP analog; nucleates and stabilizes short microtubules. | Jena Bioscience; used to prevent dynamic instability and annealing [2]. |
| MAP65 / PRC1 / Ase1 | Antiparallel microtubule crosslinking protein; defines bipolar tactoid shape. | Recombinant protein (e.g., from plant MAP65-1, human PRC1) [2] [16]. |
| PEG (Polyethylene Glycol) | Macromolecular crowding agent; induces depletion-induced bundling. | Common molecular weights: 5-20 kDa [17] [2]. |
| PIPES Buffer | Standard buffer for tubulin and microtubule experiments; maintains pH. | 80 mM PIPES, pH 6.8-7.0, with 1 mM EGTA, 1-2 mM MgCl₂ [19]. |
| Taxol / Paclitaxel | Microtubule-stabilizing drug; used in some protocols to suppress dynamics. | Cytoskeleton Inc.; often used at low micromolar concentrations [19]. |
| Silanized Coverslips | Hydrophobic surface for subsequent polymer brush coating to prevent adhesion. | Prepared in-lab with dimethyldichlorosilane [2]. |
Signaling Pathways and Experimental Workflows
Molecular Regulation of the Ase1/PRC1/MAP65 Family
The following diagram summarizes the key regulatory inputs that control the activity of the central crosslinkers in this family, such as PRC1, during cell division.
Diagram 1: Regulatory network of PRC1 family proteins.
Workflow for Microtubule Tactoid Self-Assembly
This flowchart outlines the major experimental steps for successfully creating and analyzing microtubule tactoids, from reagent preparation to data collection.
Diagram 2: Microtubule tactoid self-assembly workflow.
The Role of Short, Stable Microtubules and Antiparallel Crosslinking in Shape Determination
The self-organization of microtubules into specific, functional architectures is a fundamental process in cell division, polarity, and motility. Central to this process is the formation of the mitotic spindle, a structure that shares remarkable geometric similarity with liquid crystal tactoids. These spindle-like assemblies can be reconstituted in vitro using a minimal set of components: short, stable microtubules and specific antiparallel microtubule crosslinkers from the MAP65/PRC1/Ase1 family [20] [1]. The interplay between microtubule length and crosslinking activity is critical for determining the final shape and mechanical properties of these cytoskeletal structures. This application note details the quantitative relationships and experimental protocols that underpin the role of these components in shape determination, providing a resource for researchers aiming to reconstitute and study these architectures.
Core Principles: Geometry and Crosslinking in Shape Determination
The Mechanistic Role of Antiparallel Crosslinkers
Proteins such as PRC1 (in mammals), Ase1 (in yeast), and MAP65 (in plants) are evolutionarily conserved non-motor microtubule-associated proteins (MAPs) that selectively crosslink microtubules in an antiparallel orientation [21]. They serve as the primary architectural element that "marks" regions of antiparallel overlap, forming compliant crosslinks that define bundle organization.
Structural and biophysical studies reveal that PRC1 functions as a homodimer, utilizing a combination of a structured spectrin-fold domain and an unstructured Lys/Arg-rich basic domain to bind microtubules [21]. This dual-domain structure is key to its function. The linker between the two microtubule-binding domains exhibits flexibility on single microtubules but forms well-defined, rigid crossbridges when engaging two antiparallel filaments [21]. This unique property allows PRC1 to selectively stabilize antiparallel configurations while allowing for dynamic reorganization.
The Influence of Microtubule Length on Assembly Geometry
The length of the microtubule building blocks is a critical determinant of the final assembly's shape. Long microtubules tend to form extended bundles. In contrast, short microtubules are essential for the formation of tapered, spindle-shaped tactoids [20]. The use of GMPCPP, a non-hydrolyzable GTP analog, to nucleate and stabilize short filaments is a common strategy to obtain a high density of short microtubules that serve as the mesogens for tactoid self-assembly [20].
Table 1: Quantitative Impact of Geometrical Parameters on Microtubule Organization by PRC1 and Kif4A
| Geometrical Parameter | Experimental System | Impact on Microtubule Organization | Quantitative Relationship |
|---|---|---|---|
| Initial Overlap Length | PRC1 & Kinesin-4 (Kif4A) [22] | Regulates sliding velocity | Sliding velocity scales with initial microtubule-overlap length |
| Microtubule Length | PRC1 & Kinesin-4 (Kif4A) [22] | Defines size of final stable overlap | Width of the final overlap scales with microtubule lengths |
| Motor Density | Kinesin-14 (Ncd) [23] | Determines rotational pitch of sliding microtubules | Helical pitch varies with motor density (0.5 to 3 µm observed) |
| Motor Extension | Kinesin-14 (Ncd) [23] | Defines spatial separation of cross-linked filaments | In situ motor extension measured at ~20 nm |
Key Experimental Protocols
Protocol 1: Formation of Microtubule Tactoids
This protocol enables the self-assembly of spindle-shaped microtubule tactoids using the antiparallel crosslinker MAP65 and a crowding agent [20] [1].
Reagents and Materials:
- Tubulin: Purified, lyophilized, unlabeled and fluorescently-labeled.
- Crosslinker: MAP65, Ase1, or PRC1.
- Crowding Agent: Polyethylene Glycol (PEG).
- Stabilizing Nucleotide: GMPCPP for nucleating short, stable microtubules.
- Imaging Chamber Components: Silanized coverslips.
Procedure:
- Coverslip Silanization: Clean coverslips are irradiated with UV-Ozone to remove background fluorescence. They are then treated with dimethyldichlorosilane (DDS) in a fume hood to create a hydrophobic surface, which facilitates subsequent coating with a block copolymer to form a polymer brush [20].
- Tubulin Preparation: Resuspend lyophilized tubulin in cold PEM-80 buffer (80 mM PIPES, 1 mM MgCl₂, 1 mM EGTA, pH 6.8). Mix unlabeled and fluorescently-labeled tubulin to a final concentration suitable for visualization (e.g., 5 mg/mL) [20].
- Short Microtubule Preparation: Polymerize tubulin in the presence of GMPCPP to generate a population of short, stable microtubules. The specific incubation conditions (time, temperature) will determine the average filament length.
- Tactoid Assembly: In the assembled imaging chamber, combine the short microtubules with MAP65 and PEG. The final solution should contain:
- Microtubules (concentration tuned for assembly, e.g., 2-5 µM tubulin dimer)
- MAP65/PRC1 (e.g., 50-100 nM)
- PEG (e.g., 1-2% w/v as a crowding agent)
- Incubation and Imaging: Allow the mixture to incubate for 15-60 minutes at room temperature to facilitate self-assembly. Image the resulting tactoids using fluorescence microscopy. Characterize shape and constituent mobility using techniques like Fluorescence Recovery After Photobleaching (FRAP) [20] [1].
Protocol 2: Simultaneous Visualization of Crosslinked and Single Microtubule Dynamics
This TIRF microscopy assay allows for the direct comparison of dynamic properties between PRC1-crosslinked microtubule bundles and single microtubules under identical conditions [24].
Reagents and Materials:
- Assay Buffer (BRB80): 80 mM PIPES, 1 mM MgCl₂, 1 mM EGTA, pH 6.8.
- Oxygen Scavenging System: Glucose oxidase, catalase, and glucose to reduce photodamage.
- Blocking Agents: Kappa-casein or Bovine Serum Albumin (BSA).
- Microtubule Seeds: Biotinylated, GMPCPP-stabilized microtubule fragments for surface immobilization.
Procedure:
- Imaging Chamber Preparation: Create a flow chamber using a silanized coverslip and a glass slide. Introduce NeutrAvidin (0.2 mg/mL) to coat the surface, followed by a blocking step with kappa-casein (0.5 mg/mL) [24].
- Surface Immobilization of Microtubule Seeds: Flow in biotinylated microtubule seeds and allow them to immobilize on the NeutrAvidin-coated surface.
- Generation of Crosslinked Bundles: Introduce PRC1 (e.g., 0.2 nM) to form crosslinks between immobilized seeds and free microtubules, creating bundled architectures alongside single microtubules [22] [24].
- Initiation of Dynamics: Flow in the final assay mixture containing:
- Soluble tubulin (e.g., 10-15 µM)
- GTP (1 mM)
- ATP (1 mM, if motors are used)
- Oxygen scavenging system
- Relevant MAPs or motor proteins of interest
- Data Acquisition and Analysis: Use multi-wavelength TIRF microscopy to simultaneously record the dynamics (growth, shrinkage, catastrophe, rescue) of single and crosslinked microtubules over time. Kymograph analysis is used to extract dynamic parameters [24].
The following workflow diagram illustrates the key steps for this protocol:
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Microtubule Crosslinking and Shape Determination Studies
| Research Reagent | Function in Experiment | Key Characteristics & Examples |
|---|---|---|
| Antiparallel Crosslinkers | Selectively binds and bundles antiparallel microtubules, defining array architecture. | PRC1 (human), Ase1 (yeast), MAP65 (plant); homo-dimeric [21]. |
| Kinesin Motor Proteins | Drives relative sliding and generates mechanical forces within crosslinked arrays. | Kinesin-4 (Kif4A): plus-end-directed, regulates sliding and dynamics [22]. Kinesin-14 (Ncd): minus-end-directed, induces helical sliding [23]. |
| Microtubule Stabilizers | Generates short, stable microtubules as building blocks for defined assemblies. | GMPCPP: nucleates short, stable filaments resistant to catastrophe [20]. Taxol: stabilizes microtubule lattice, suppresses dynamics. |
| Crowding Agents | Mimics intracellular crowded environment, promotes condensation and phase separation. | Polyethylene Glycol (PEG): induces depletion forces that bundle filaments [20]. Methylcellulose: increases solution viscosity, used in motility assays. |
| Actin-Microtubule Crosslinkers | Links actin and microtubule cytoskeletons, enabling crosstalk and force transmission. | Tau: MAP that bundles F-actin and crosslinks it to microtubules [25]. TipAct: engineered crosslinker for actin transport by microtubule ends [26]. |
Advanced Concepts and Inter-System Crosstalk
The principles of shape determination extend beyond single-network systems. A powerful example of cytoskeletal crosstalk is the transport of actin filaments by growing microtubule ends, mediated by passive crosslinkers like the engineered TipAct protein [26]. This transport arises from a force balance: a forward condensation force is generated by the preferential binding of crosslinkers to the chemically distinct microtubule tip region, which attempts to maximize the overlap with the actin filament. This is antagonized by a backward friction force caused by crosslinkers binding to the microtubule lattice [26]. This mechanism, distinct from motor-driven transport, illustrates how passive crosslinkers can enable one cytoskeletal system to actively remodel another.
The following diagram illustrates this force balance mechanism:
Data Presentation and Analysis
Quantitative analysis is key to understanding the mechanisms of shape determination. The following parameters should be measured and analyzed:
- For Tactoid Assays:
- Shape Characterization: Length, width, and aspect ratio of assembled tactoids.
- Material Properties: FRAP to measure mobility and turnover of constituents (tubulin, crosslinkers) within the assembly [20].
- For Sliding and Dynamics Assays:
- Sliding Velocity: Tracked from kymographs or time-lapse sequences [22] [24].
- Helical Parameters: For 3D assays, measure helical pitch and diameter of sliding trajectories [23].
- Dynamic Instability Parameters: For dynamic microtubules, quantify growth/shrinkage rates, and catastrophe/rescue frequencies from single and crosslinked populations [24].
The integration of short, stable microtubules with specific antiparallel crosslinking proteins provides a powerful minimal system for understanding the physical principles of cytoskeletal shape determination. The protocols and data outlined here offer a reproducible path for in vitro reconstitution and quantitative analysis, forming a critical foundation for research in cell biophysics, cytoskeletal engineering, and drug discovery targeting cellular architecture.
A Detailed Protocol for Creating and Observing Microtubule Tactoids
Research Reagent Solutions
The following table details the essential reagents and materials required for research on tubulin and microtubule tactoid self-assembly.
| Item Name | Function & Application |
|---|---|
| Tubulin (from brain tissue) | The fundamental protein subunit (dimer) that polymerizes to form microtubules. Purified from fresh porcine or bovine brain via polymerization/depolymerization cycles. [27] [28] |
| MAP65/Ase1/PRC1 | An antiparallel microtubule crosslinking protein, essential for bundling microtubules into spindle-like tactoid structures. A key minimal component for in vitro reconstitution. [2] [15] |
| GMPCPP (Guanosine-5'-[(α,β)-methyleno]triphosphate) | A slowly-hydrolyzable GTP analog. Used to nucleate and stabilize microtubules, preventing dynamic instability and enabling the creation of short, stable microtubules necessary for tactoid formation. [29] [2] |
| PEG (Polyethylene Glycol) | A crowding agent that creates depletion interactions, increasing the local concentration of microtubules and promoting their condensation and self-assembly into larger structures like tactoids. [2] |
| GTP (Guanosine Triphosphate) | The natural nucleotide required for tubulin polymerization. It is incorporated into the growing microtubule end and subsequently hydrolyzed, regulating dynamics. [27] [29] |
| Phosphocellulose (PC) Resin | A cation-exchange chromatography medium used for the final purification step of tubulin, separating it from microtubule-associated proteins (MAPs). [27] [28] |
| Pipes Buffer | A common buffering agent (pH 6.6-6.9) used in tubulin and microtubule protocols to maintain a stable chemical environment. [27] |
Tubulin Preparation from Brain Tissue
This protocol describes the large-scale preparation of tubulin from fresh pig or cow brains, adapted from established methods. [27] [28]
Materials and Equipment
- Biological Material: 3-5 fresh pig brains or a larger quantity (e.g., 10) of fresh cow brains.
- Buffers:
- PM Buffer: 100 mM Pipes (pH 6.9), 2 mM EGTA, 1 mM MgSO₄, 2 mM DTT. [27]
- PM-4M Buffer: PM Buffer with 4 M Glycerol. [27]
- PM-8M Buffer: PM Buffer with 8 M Glycerol. [27]
- PB (Pipes/Polymerization Buffer): 0.1 M K-Pipes (pH 6.8), 0.5 mM MgCl₂, 2 mM EGTA, 0.1 mM EDTA, 0.1% β-mercaptoethanol, 1 mM ATP. [28]
- Nucleotides: GTP and ATP. [27] [28]
- Key Equipment: Waring blender, Dounce homogenizer, high-speed centrifuge (e.g., Sorvall with SS-34 rotor), ultracentrifuge (e.g., Beckman with Ti70 rotor), phosphocellulose (PC) column. [27] [28]
Protocol Steps
- Homogenization: Remove meninges and blood vessels from fresh brains in a cold room. Homogenize the tissue in a pre-cooled Waring blender with PM-4M buffer. [27]
- Clarification: Centrifuge the homogenate at low speed (e.g., 9,500 rpm in an SS-34 rotor) to remove debris. Then, centrifuge the resulting supernatant at high speed (e.g., 96,000 × g) to obtain a clear lysate. [27]
- First Polymerization Cycle: Add GTP to the lysate to a final concentration of 0.5 mM. Incubate the mixture at 34-37°C for 45-60 minutes to polymerize microtubules. Pellet the polymerized microtubules by warm centrifugation (e.g., 30,000 rpm at 27°C). [27] [28]
- First Depolymerization: Resuspend the pellet in a small volume of cold PM buffer and homogenize using a Dounce homogenizer. Incubate on ice for 30 minutes to depolymerize microtubules. Clarify by cold, high-speed centrifugation to remove aggregates. The supernatant contains tubulin. [27]
- Second Polymerization/Depolymerization Cycle: Repeat steps 3 and 4 to increase the purity of the tubulin. The volume of buffer used for resuspension can be based on the volume from the first cycle (e.g., 0.25 × V1). [27]
- Phosphocellulose Chromatography: Apply the twice-cycled tubulin supernatant to a pre-equilibrated PC column. Elute with PM buffer. The flow-through contains pure tubulin, while microtubule-associated proteins (MAPs) bind to the column. [27]
- Storage: Pool the pure tubulin fractions, add an equal volume of PM-8M buffer, divide into aliquots, freeze in liquid nitrogen, and store at -80°C. [27]
Self-Assembly of Microtubule Tactoids
This protocol describes the formation of spindle-shaped microtubule assemblies (tactoids) using stabilized microtubules, the crosslinker MAP65, and PEG. [2]
Materials
- Proteins: Purified tubulin, MAP65.
- Nucleotide: GMPCPP.
- Crowding Agent: Polyethylene Glycol (PEG).
- Imaging Supplies: Glass coverslips.
- Specialized Chemicals: Dimethyldichlorosilane (DDS) for silanization. [2]
Protocol Steps
- Coverslip Silanization (for sample chambers):
- Clean coverslips with water and ethanol, then treat with UV-Ozone (UVO) or plasma to remove organic residue. [2]
- In a fume hood, immerse the coverslips in a 2% solution of dimethyldichlorosilane (DDS) in a suitable container for 5 minutes. Caution: DDS is highly toxic. [2]
- Rinse the coverslips thoroughly with 100% ethanol and then water. Air-dry completely in the fume hood. This creates a hydrophobic surface for subsequent polymer brush coating. [2]
- Formation of GMPCPP-stabilized Microtubules:
- Polymerize tubulin in the presence of GMPCPP to create short, stabilized microtubules. The specific protocol for GMPCPP microtubule growth should be followed. [2]
- Tactoid Assembly Reaction:
- Combine the stabilized, short microtubules with the antiparallel crosslinker MAP65.
- Include PEG in the reaction mixture to act as a crowding agent, which promotes self-assembly through depletion forces. [2]
- Incubate the mixture to allow for the formation of long, thin, spindle-shaped tactoids.
- Characterization:
Table 1: Key Buffers for Tubulin Preparation
| Buffer Name | Composition | Primary Function |
|---|---|---|
| PM Buffer [27] | 100 mM Pipes (pH 6.9), 2 mM EGTA, 1 mM MgSO₄, 2 mM DTT | General tubulin handling and depolymerization. |
| PM-4M Buffer [27] | PM Buffer + 4 M Glycerol | Homogenization buffer; glycerol aids in polymerization cycles. |
| PM-8M Buffer [27] | PM Buffer + 8 M Glycerol | Tubulin storage at -20°C. |
| PB (Pipes/Polymerization Buffer) [28] | 0.1 M K-Pipes (pH 6.8), 0.5 mM MgCl₂, 2 mM EGTA, 0.1 mM EDTA, 0.1% β-mercaptoethanol, 1 mM ATP | Buffer used in large-scale preps for polymerization. |
Table 2: Centrifugation Parameters for Tubulin Prep
| Step | Rotor Type | Speed & Duration | Temperature | Purpose |
|---|---|---|---|---|
| Clarification [27] | Sorvall SS-34 | 9,500 rpm for 15 min | 4°C | Remove tissue debris. |
| Lysate Clearance [27] | Beckman Ti70 | 96,000 × g for 75 min | 4°C | Obtain clear supernatant for polymerization. |
| Polymer Pellet (Cycle 1) [27] | Beckman Ti70 | 96,000 × g for 60 min | 27°C | Pellet polymerized microtubules. |
| Depolymerization Clearance [27] | Beckman Ti70 | 96,000 × g for 60 min | 4°C | Remove aggregates after depolymerization. |
Experimental Workflow Diagrams
Tubulin Protein Purification Workflow
Tactoid Self-Assembly Workflow
Nucleotide Role in Microtubule Assembly
Within the broader research on the self-assembly of microtubule tactoids, the preparation of the experimental substrate is a critical foundational step. The formation of these spindle-like liquid crystal assemblies, which serve as minimalistic models for the mitotic spindle, is highly sensitive to surface interactions [20] [7]. The self-organization of microtubules into tactoids requires a specific set of conditions, including the use of an antiparallel crosslinker like MAP65 and a crowding agent such as Polyethylene Glycol (PEG) [20] [2]. The role of the flow chamber surface is to minimize undesired adsorption of these components and to prevent non-specific sticking of microtubules, which could disrupt the delicate self-organization process [20]. The protocol outlined herein describes the silanization of glass coverslips to create a hydrophobic surface, which subsequently allows for the application of a non-adsorbing polymer brush coating (e.g., Pluronic F-127) within the final flow chamber [20] [7]. This treatment is essential for achieving a reproducible environment in which microtubule tactoids can form and be observed without interference from the surface.
Materials and Reagents
Research Reagent Solutions
The following table details the key materials required for the silanization process and their specific functions in the protocol.
Table 1: Essential Materials for Coverslip Silanization
| Item Name | Function/Explanation |
|---|---|
| Glass Coverslips | Serves as the primary substrate for constructing the flow chamber. |
| Dimethyldichlorosilane (DDS) | A highly toxic silane compound that reacts with the glass surface to create a stable hydrophobic layer [20]. This layer is crucial for the subsequent attachment of a block copolymer brush. |
| Acetone (100%) | Organic solvent used for deep cleaning the coverslip surface to remove organic residues [20]. |
| Ethanol (100%) | Used for rinsing and cleaning steps to remove water and other solvents [20]. |
| Potassium Hydroxide (KOH), 0.1 M | Aqueous base used to clean and hydroxylate the glass surface, preparing it for the silanization reaction [20]. |
| UV-Ozone (UVO) Machine or Plasma Chamber | Used for high-level cleaning and removal of any fluorescent contaminants from the glass surface prior to silanization [20]. |
Equipment
- Metal coverslip-holding racks (dedicated one for UVO/plasma and another for silanization)
- Fume hood
- Lint-free laboratory wipes
- Tweezers
- Containers for immersion steps
Experimental Protocol
NOTE: The following procedure must be performed in a fume hood while wearing appropriate personal protective equipment, including gloves. Dimethyldichlorosilane is highly toxic and must be handled with utmost care [20].
Initial Coverslip Cleaning
- Rinse: Rinse the coverslips thoroughly with ddH₂O, followed by 70% ethanol, and then ddH₂O again. Dry the coverslips with lint-free laboratory wipes between each rinse. This sequence removes dust and water-soluble or organic particles from the surface [20].
- UV-Ozone/Plasma Treatment: Place the coverslips in a dedicated metal rack and transfer them to a UV-Ozone (UVO) machine. Irradiate the coverslips for 20 minutes to eliminate any background fluorescence. A plasma chamber can be used as an effective alternative to UVO [20].
- Transfer: Using tweezers, transfer the coverslips from the rack used for UVO treatment to a different, pre-cleaned metal rack designated for the silanization process. Using separate racks prevents cross-contamination and high levels of oxidation [20].
Detailed Solvent Cleaning Sequence
This series of immersion steps ensures the surface is impeccably clean and ready for chemical modification.
- Acetone Immersion: Immerse the rack with coverslips in a container with 100% acetone for 1 hour. After immersion, rinse the container 3 times with tap water and then 3 times with ddH₂O to remove all traces of acetone [20].
- Ethanol Immersion: Immerse the rack in 100% ethanol for 10 minutes. Rinse the container 3 times with tap water and then 3 times with ddH₂O [20].
- Water Rinses: Immerse the rack 3 times in ddH₂O for 5 minutes each [20].
- KOH Immersion: Immerse the rack in 0.1 M KOH (prepared by adding 50 mL of 1 M KOH to 450 mL of ddH₂O) for 15 minutes. Rinse the container 3 times with tap water and then 3 times with ddH₂O [20].
- Final Water Rinses: Immerse the rack 3 more times in ddH₂O for 5 minutes each [20].
- Drying: Air-dry the rack with the coverslips overnight in a fume hood or laminar flow hood [20].
Silanization Reaction
- Silane Application: After ensuring the coverslips and rack are completely dry, immerse them for 5 minutes in a 2% solution of dimethyldichlorosilane (DDS) in a container used specifically for silane. It is critical that no moisture is introduced at this stage. [20]
- Ethanol Rinses: Immerse the rack and coverslips 2 times in a container with 100% ethanol for 5 minutes each. Rinse the container 3 times with tap water and then 3 times with ddH₂O afterward [20].
- Water Rinses: Immerse the rack and coverslips 3 times in ddH₂O for 5 minutes each [20].
- Final Drying: Air-dry the rack with the coverslips overnight in a fume hood or laminar flow hood [20].
- Storage: Using tweezers, transfer the silanized coverslips into coverslip boxes. These treated coverslips remain usable for 1-2 months. Coverslips that are stored longer may lose their coating effectiveness and should be discarded [20].
Workflow Visualization
The following diagram illustrates the sequential stages of the flow chamber preparation process, from initial cleaning to the final readiness for the tactoid assay.
Quantitative Data & Specifications
Table 2: Critical Parameters and Timing for the Silanization Protocol
| Process Step | Key Parameter | Specification / Value | Notes |
|---|---|---|---|
| UV-Ozone Cleaning | Duration | 20 minutes | Alternatively, use a plasma chamber. |
| Acetone Immersion | Duration & Concentration | 1 hour in 100% Acetone | Removes organic residues. |
| KOH Immersion | Concentration & Duration | 15 minutes in 0.1 M KOH | Cleans and hydroxylates the surface. |
| Silanization | Concentration & Duration | 5 minutes in 2% DDS | Critical step. Must be performed in a fume hood with dry materials. |
| Drying Steps | Duration | Overnight (x2) | Essential for complete reaction and evaporation of solvents. |
| Coated Coverslip Shelf Life | Usability Period | 1 to 2 months | Old coatings lose effectiveness and should be discarded. |
Within the broader research on the self-assembly of microtubule tactoids, the preparation of the tubulin mix and the final reaction solution constitutes a foundational and critical step. This protocol details the precise methodology for combining purified tubulin with essential nucleotides, crowding agents, and crosslinking proteins to create the conditions necessary for the spontaneous formation of spindle-shaped microtubule assemblies [2] [6]. The reproducibility of tactoid self-assembly is highly dependent on the accuracy and timing of these preparatory steps, which are designed to create a minimalistic in vitro system that mimics the liquid crystal properties of the mitotic spindle [1] [2]. The following sections provide a detailed, step-by-step guide for researchers to prepare the tubulin mix and the final reaction solution, ensuring consistent and reliable results.
Research Reagent Solutions
The following table catalogues the key reagents required for preparing the tubulin mix and reaction solution for microtubule tactoid assembly.
- Table 1: Essential Reagents for Tubulin Tactoid Assembly
Reagent Name Function in the Protocol Key Specifications / Notes Unlabeled Tubulin Primary structural protein for microtubule assembly; forms the bulk of the tactoid structure. Lyophilized, stored at -80°C. Resuspended in cold PEM-80 buffer [7]. Rhodamine-Labeled Tubulin Fluorescent marker for visualization of microtubules via fluorescence microscopy. Lyophilized, stored at -80°C. Typically constitutes a small fraction of the total tubulin mix [7]. PEM-80 Buffer Tubulin resuspension and reaction buffer. Provides the ionic conditions necessary for tubulin stability and polymerization. Composition: 80 mM PIPES, 1 mM EGTA, 1 mM MgCl₂, pH 6.8 [7]. GMPPCPP (Guanylyl-(α,β)-methylene-diphosphonate) Non-hydrolyzable GTP analogue used to nucleate and stabilize microtubule seeds, protecting them from depolymerization [2]. Creates stable microtubule nuclei from which dynamic microtubules can grow [2]. MAP65 (with GFP tag) Antiparallel microtubule crosslinker; essential for bundling microtubules into spindle-shaped tactoids. Plant homolog of PRC1/Ase1. GFP tag allows visualization of the crosslinker's localization [2] [7] [6]. Polyethylene Glycol (PEG) Crowding agent. Creates depletion forces that increase the effective tubulin concentration and promote microtubule condensation [2]. --- Pluronic F-127 Non-ionic block copolymer surfactant. Passivates the chamber surface to prevent non-specific adhesion of microtubules [7]. --- Glucose Oxidase/Catalase Oxygen-scavenging system. Reduces photodamage during fluorescence microscopy by removing reactive oxygen species [7]. --- Dithiothreitol (DTT) Reducing agent. Helps maintain protein stability and function by preventing oxidation of cysteine residues [7]. ---
Detailed Methodology
Preparation of the Tubulin Working Mix
The first critical stage involves creating a ready-to-use tubulin aliquot that combines unlabeled and fluorescently labeled tubulin.
- Thaw Reagents: Retrieve one aliquot of unlabeled tubulin (1 mg lyophilized powder) and one aliquot of rhodamine-labeled tubulin (20 µg lyophilized powder) from the -80°C freezer. Place both immediately on ice [7].
- Resuspend Tubulin:
- To the 1 mg unlabeled tubulin aliquot, add 200 µL of cold PEM-80 buffer.
- To the 20 µg rhodamine-labeled tubulin aliquot, add 4 µL of cold PEM-80 buffer.
- Keep both tubes on ice for 10 minutes to ensure the lyophilized powder is fully dissolved. Gently pipette up and down if necessary, avoiding bubble formation [7].
- Combine Tubulins: Transfer 100 µL of the resuspended unlabeled tubulin solution into the tube containing the 4 µL of rhodamine-labeled tubulin [7].
- Mix and Aliquot:
- Mix the combined solutions by pipetting slowly up and down 6-7 times.
- Distribute the final tubulin mix into seven new tubes, with 15 µL in each.
- Immediately snap-freeze the aliquots in liquid nitrogen and return them to the -80°C freezer for future use. This step ensures tubulin stability and allows for reproducible experiments over time [7].
Preparation of the Final Reaction Solution
The assembly of the final reaction must be performed efficiently and with reagents kept on ice to preserve tubulin functionality. The entire process from mixing to loading into the flow chamber should be completed within 10-12 minutes [7].
- Prepare Flow Chamber: Prior to preparing the reaction mix, coat a silanized flow chamber with 20 µL of a 5% solution of Pluronic F-127 in PEM-80. Incubate the chamber in a humidified environment (e.g., a Petri dish with a wet lint-free wipe) for at least 5-7 minutes to allow for surface passivation [7].
- Combine Reaction Components: In a sterile tube kept on ice, combine the following reagents in the order listed. Mix by pipetting 5-6 times after addition [7].
- PEM-80 buffer
- GMPPCPP
- Pluronic F-127
- Dithiothreitol (DTT)
- Glucose
- Polyethylene Glycol (PEG)
- The prepared Tubulin Mix (from step 3.1)
- MAP65 (with GFP-MAP65 for visualization)
- Add Oxygen Scavengers: Introduce 1 µL of a pre-mixed glucose oxidase and catalase solution into the reaction tube. Mix the entire solution thoroughly by pipetting 7-8 times [7].
- Load the Chamber:
- Remove the Pluronic F-127 solution from the flow chamber by capillary action, using a lint-free wipe placed at the opposite end of the chamber.
- Immediately add the prepared tubulin-MAP65 reaction mix to the chamber entrance to draw it in via capillary action [7].
- Seal and Incubate: Once the sample has filled the chamber, seal both ends with five-minute epoxy glue. Transfer the chamber to a 37°C incubator for 30 minutes to allow for microtubule nucleation and tactoid growth [7].
The preparation of reagents is characterized by specific volumetric and concentration parameters, which are summarized below for quick reference.
- Table 2: Quantitative Data for Tubulin and Reaction Preparation
Parameter Value Relevance / Context Unlabeled Tubulin Mass 1 mg Mass in one primary aliquot for resuspension [7]. Rhodamine-Tubulin Mass 20 µg Mass in one primary aliquot for resuspension; creates a labeled fraction for imaging [7]. Final Tubulin Mix Aliquot Volume 15 µL Volume of single-use working aliquots stored at -80°C [7]. Chamber Coating Volume 20 µL Volume of 5% Pluronic F-127 used to passivate the flow chamber [7]. Chamber Coating Time 5-7 minutes Minimum incubation time for surface passivation [7]. Reaction Preparation Timeframe 10-12 minutes Maximum recommended time from mixing to loading; critical for tubulin activity [7]. Tactoid Assembly Incubation 30 minutes Time required at 37°C for complete tactoid formation [7]. Incubation Temperature 37°C Temperature for microtubule nucleation and tactoid growth [7].
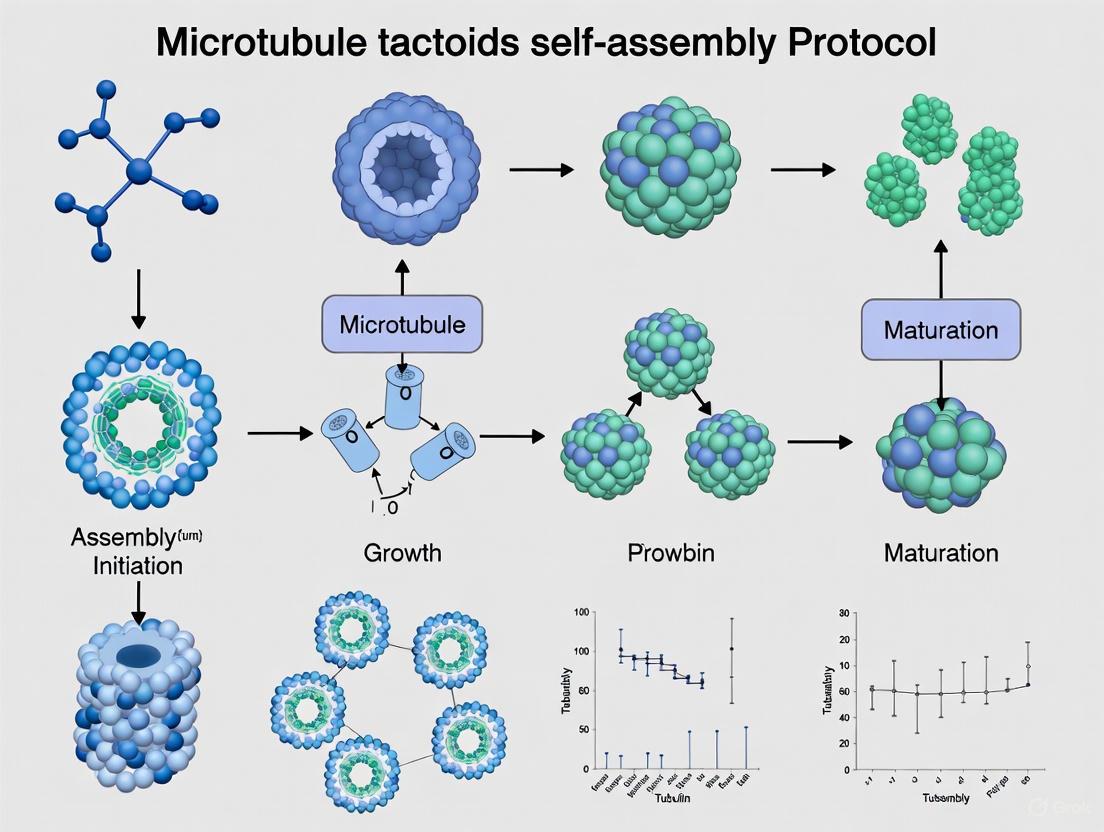
This application note details the procedure for assembling and incubating microtubule tactoids, which are spindle-shaped, liquid-crystal-like assemblies of microtubules. This protocol is a critical step in reconstituting self-organized spindle-like structures in vitro using a minimal system comprising short, stabilized microtubules, the antiparallel microtubule crosslinker MAP65, and the crowding agent polyethylene glycol (PEG) [2] [1]. The formation of these tactoids provides a valuable model for studying the physical principles of biological self-organization, relevant to processes such as mitotic spindle formation and the behavior of mesoscale liquid crystals [2] [30].
Reagent Preparation
Tubulin Stock Solution
| Component | Specification | Function |
|---|---|---|
| Unlabeled Tubulin | 1 mg lyophilized [7] | Primary structural component for microtubule assembly. |
| Rhodamine-Labeled Tubulin | 20 µg lyophilized [7] | Fluorescent marker for microtubule visualization. |
| PEM-80 Buffer | Cold [7] | Buffer for reconstituting and storing tubulin. |
Procedure:
- Obtain an aliquot of unlabeled tubulin (1 mg lyophilized) and an aliquot of rhodamine-labeled tubulin (20 µg lyophilized) from a -80 °C freezer and keep them on ice [7].
- Add 200 µL of cold PEM-80 to the unlabeled tubulin vial. Add 4 µL of cold PEM-80 to the rhodamine-labeled tubulin vial [7].
- Keep both tubes on ice for 10 minutes to dissolve the lyophilized material completely [7].
- Combine 100 µL of the resuspended unlabeled tubulin with the entire 4 µL of rhodamine-labeled tubulin. Mix by pipetting slowly 6-7 times [7].
- Distribute the tubulin mix into aliquots of 15 µL in new tubes. Flash-freeze the aliquots in liquid nitrogen and store them at -80 °C for future use [7].
MAP65 Working Solution
| Component | Specification | Function |
|---|---|---|
| MAP65 | Plant-derived, recombinant [2] | Anti-parallel microtubule crosslinking protein, essential for tactoid shape. |
| GFP-MAP65 | Optional [7] | Fluorescently tagged version for visualizing crosslinker localization. |
Assembly Mix Components
| Component | Typical Concentration/Amount | Function |
|---|---|---|
| PEM-80 Buffer | To volume [7] | Reaction buffer. |
| GMPCPP | As required [7] | Non-hydrolyzable GTP analog; nucleates and stabilizes short microtubules. |
| Pluronic F-127 | As required [7] | Non-ionic block copolymer surfactant; prevents surface adhesion. |
| Dithiothreitol (DTT) | As required [7] | Reducing agent; maintains protein integrity. |
| Glucose | As required [7] | Substrate for oxygen-scavenging system. |
| Polyethylene Glycol (PEG) | As required [2] [7] | Macromolecular crowder; induces depletion forces that promote tactoid condensation. |
| Glucose Oxidase/Catalase Mix | 1 µL pre-mixed [7] | Oxygen-scavenging system; reduces photodamage during fluorescence imaging. |
Experimental Workflow
The following diagram illustrates the key stages of the tactoid assembly and incubation protocol.
Detailed Protocol
Flow Chamber Assembly
Principle: A sealed, passivated flow chamber is required to contain the reaction mixture and prevent non-specific adhesion of microtubules to glass surfaces, which would inhibit tactoid formation [2] [7].
Procedure:
- Silanization of Coverslips (Perform in a fume hood):
- Rinse coverslips sequentially with ddH₂O, 70% ethanol, and ddH₂O, drying with lint-free wipes between rinses [2].
- Place coverslips in a metal rack and irradiate with UV-Ozone (UVO) for 20 minutes (or use plasma treatment) to remove background fluorescence [2].
- Immerse the rack with coverslips in 2% dimethyldichlorosilane (DDS) in a dedicated container for 5 minutes. Caution: DDS is highly toxic. [2]
- Rinse the rack and coverslips twice in 100% ethanol for 5 minutes each [2].
- Air-dry the rack and coverslips overnight in a fume hood or laminar flow hood [2].
- Chamber Construction:
- Clean a glass slide with ddH₂O and ethanol, then dry it [7].
- Create two thin strips from double-sided tape (40-50 mm long) and place them on the slide 5-8 mm apart to form a flow path [7].
- Place a silanized coverslip on top of the tape strips to form the chamber [7].
- Press gently with the back of a pen on the tape region to seal. A good seal will turn the tape from translucent to clear [7].
- Trim excess tape with a razor blade, leaving about 1 mm at the chamber entrance [7].
Tactoid Assembly and Incubation
Principle: The core assembly mix combines stabilized microtubule seeds, free tubulin, the MAP65 crosslinker, and a crowding agent. Under controlled incubation, this leads to the self-organization of microtubules into tactoids [2] [7].
Procedure:
- Thaw all necessary reagents (tubulin aliquot, MAP65, etc.) on ice [7].
- Chamber Coating: Pipette 20 µL of 5% Pluronic F-127 surfactant in PEM-80 into the flow chamber. Place the chamber in a humidified Petri dish for at least 5-7 minutes [7].
- Prepare Assembly Mix: In a sterile tube on ice, combine the following by pipetting 5-6 times:
- PEM-80 buffer
- GMPCPP
- Pluronic F-127
- Dithiothreitol (DTT)
- Glucose
- Polyethylene Glycol (PEG)
- Thawed tubulin mix
- MAP65 (including a small amount of GFP-MAP65 for visualization) [7].
- Add 1 µL of the pre-mixed glucose oxidase/catalase solution to the assembly mix. Mix by pipetting 7-8 times [7].
- Critical Note: The entire process of preparing the assembly mix should be completed within 10-12 minutes to maintain tubulin integrity [7].
- Load Chamber: Remove the Pluronic F-127 solution from the flow chamber by capillary action using a lint-free wipe at one end. Immediately add the tubulin-MAP65 assembly mix to the other end of the chamber [7].
- Once the sample has filled the chamber, seal both ends with five-minute epoxy [7].
- Incubate: Transfer the sealed chamber to a 37°C incubator for approximately 30 minutes to allow for microtubule nucleation and tactoid growth [7].
Expected Results and Quality Control
After a 30-minute incubation at 37°C, microtubule tactoids should be visible under fluorescence microscopy [7]. These structures are typically spindle-shaped and should be visible in both the tubulin channel (using a 561 nm laser for rhodamine) and the MAP65 channel (using a 488 nm laser for GFP), with the signals overlapping perfectly [7]. The formation of these tactoids confirms a successful experiment.
Characterization and Analysis
Post-incubation, tactoids can be characterized using the following methods:
| Method | Key Parameter Measured | Technical Specification |
|---|---|---|
| Fluorescence Microscopy | Tactoid morphology (length, width), co-localization of microtubules and MAP65 [2] [7]. | Objective: 60x or higher, NA ≥1.2. Camera: CMOS or CCD. Save as 16-bit TIFF [7]. |
| Fluorescence Recovery After Photobleaching (FRAP) | Mobility of constituents within the tactoid [2]. | Microtubules in tactoids are typically immobile (no recovery), while MAP65 is mobile (shows fluorescence recovery) [7]. |
Troubleshooting Guide
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| No tactoids form | Tubulin degraded on ice | Work quickly; prepare and load the assembly mix within 10-12 minutes [7]. |
| Incorrect MAP65 activity | Verify protein quality and concentration. | |
| Insufficient crowding | Ensure PEG is fresh and concentration is correct. | |
| Tactoids are not spindle-shaped | Microtubules are too long | Use GMPCPP to ensure a high density of short, stable microtubules [2] [14]. |
| Poor fluorescence signal | Low laser power or camera saturation | Adjust illumination power and exposure time to avoid saturation while maintaining a good signal [7]. |
The Scientist's Toolkit: Research Reagent Solutions
| Essential Material | Function in the Protocol |
|---|---|
| GMPCPP-stabilized Microtubules | Provides a high density of short, non-dynamic microtubule seeds that act as mesogens for liquid crystal tactoid formation [2] [14]. |
| MAP65 / PRC1 / Ase1 | Anti-parallel microtubule crosslinking protein; essential for organizing microtubules into the specific bipolar, spindle-like shape of tactoids [2] [1]. |
| PEG (Macromolecular Crowder) | Induces depletion attraction between microtubules, promoting their condensation and phase separation into tactoids [2] [7]. |
| Pluronic F-127 | A non-ionic block copolymer surfactant; coats glass surfaces to create a non-adhesive passivation layer, preventing microtubules from sticking and allowing free self-organization [7]. |
| Glucose Oxidase/Catalase System | An oxygen-scavenging system that mitigates photodamage and preserves fluorescence during prolonged microscopy imaging [7]. |
The analysis of microtubule tactoids, which are spindle-shaped liquid crystalline assemblies, is a crucial part of research on cytoskeletal self-organization. These structures serve as in vitro models for biological systems such as the mitotic spindle [20] [6]. Fluorescence microscopy provides a powerful method for visualizing these assemblies and characterizing their morphology and internal dynamics. This protocol details the specific imaging procedures for detecting microtubule tactoids using both GFP and Rhodamine channels, allowing for simultaneous observation of the structural framework formed by microtubules and the spatial distribution of the cross-linking MAP65 protein [20] [7]. The data obtained from this imaging step is fundamental for understanding the self-assembly process governed by a minimal set of components, a key theme in broader thesis research on in vitro reconstitution of complex biological structures.
Research Reagent Solutions and Essential Materials
Successful imaging requires specific reagents and equipment to prepare and stabilize the samples. The table below lists the key materials used in this protocol [20] [7].
Table 1: Essential Research Reagents and Materials
| Item | Function in the Experiment |
|---|---|
| Rhodamine-labeled tubulin | Fluorescently labels microtubule filaments, enabling visualization of the overall tactoid structure in the red channel. |
| MAP65 with GFP tag | Labels the antiparallel microtubule cross-linker protein, enabling visualization of its binding and distribution within the tactoid in the green channel. |
| Pluronic F-127 | A non-ionic block copolymer surfactant used to coat the flow chamber, preventing nonspecific absorption of proteins to the glass surface. |
| Glucose Oxidase/Catalase | An oxygen-scavenging system added to the assay buffer to reduce photobleaching of fluorophores during extended microscopy. |
| Polyethylene Glycol (PEG) | A crowding agent that mimics the crowded intracellular environment, promoting depletion interactions that assist in tactoid assembly. |
| GMPCPP | A slowly hydrolysable GTP analog used to nucleate and stabilize short microtubule filaments, which is a prerequisite for tactoid formation. |
Microscope Configuration and Equipment Setup
Proper configuration of the microscope system is critical for acquiring high-quality, multi-channel data. The following equipment and settings are recommended [20] [7].
Core Microscope Hardware
- Microscope: An inverted epifluorescence or TIRF microscope, equipped with a high-numerical aperture (NA) objective lens ( ≥60x magnification with an NA of 1.2 or higher ) to collect sufficient light.
- Camera: A high-sensitivity camera, such as a cooled CCD or sCMOS camera, for detecting low-intensity fluorescence signals.
- Excitation Sources: Lasers are preferred for stable intensity output. The required laser lines are:
- A 561 nm laser for exciting Rhodamine-labeled tubulin.
- A 488 nm laser for exciting GFP-labeled MAP65.
- Environmental Chamber: A stage-top incubator or heater system to maintain the sample at a constant 37°C throughout the imaging process, which is essential for tactoid stability.
Filter Cube Specifications
For systems using filter cubes rather than laser-based TIRF, the following specifications should be used [7]:
Table 2: Filter Cube Configurations for Dual-Channel Imaging
| Fluorophore | Excitation Filter | Dichroic Mirror | Emission Filter |
|---|---|---|---|
| Rhodamine | 540/12.5 nm | 545 nm (12.5 nm cutoff) | 575 nm Long-pass |
| GFP | 480/15 nm | 505 nm (15 nm cutoff) | 515 nm Long-pass |
Detailed Experimental Imaging Protocol
This section provides a step-by-step workflow for preparing the sample and acquiring images of the microtubule tactoids.
The following diagram summarizes the key stages of the experimental workflow from sample preparation to data acquisition.
Step-by-Step Procedure
Sample Preparation and Chamber Assembly
- Thaw all pre-mixed reagents, including the tubulin mix (containing Rhodamine-labeled tubulin) and MAP65 (with GFP-MAP65), on ice.
- Assemble a flow chamber using a silanized coverslip and a glass slide, separated by double-sided tape to create a flow path [20] [7].
- Coat the flow chamber with 20 µL of a 5% solution of Pluronic F-127 in PEM-80 buffer. Incubate the chamber in a humidified environment for at least 5-7 minutes. This step passivates the glass surface to prevent protein adhesion [7].
Initiate Tactoid Formation
- Prepare the final reaction mixture on ice by combining PEM-80 buffer, GMPCPP, Pluronic F-127, an oxygen-scavenging system (glucose oxidase and catalase), PEG, the tubulin mix, and MAP65 with GFP-MAP65.
- Remove the Pluronic F-127 solution from the flow chamber by capillary action using a lint-free wipe.
- Immediately load the freshly prepared reaction mixture into the chamber.
- Seal both ends of the chamber with quick-set epoxy to prevent evaporation.
- Incubate the sealed chamber at 37°C for 30 minutes to allow for the nucleation and growth of microtubule tactoids [7].
Microscope Setup and Image Acquisition
- Place the flow chamber on the microscope stage, ensuring temperature stability at 37°C.
- Using the Rhodamine channel (561 nm laser), locate fields of view containing tactoids. These will appear as bright, spindle-shaped structures.
- Acquire images sequentially through both fluorescence channels to prevent bleed-through between signals.
- For the Rhodamine channel (Microtubules): Use the 561 nm laser with an exposure time that does not saturate the camera's pixel intensity. Ensure the power at the sample is at least 1 mW for a good signal-to-noise ratio [7].
- For the GFP channel (MAP65): Switch to the 488 nm laser and acquire an image of the same field of view with appropriate exposure settings.
- Acquire at least 10 images from different areas of the sample to capture data on over 100 individual tactoids, ensuring statistical robustness [7].
- Save all images in a 16-bit TIFF format to preserve the dynamic range of the data for subsequent quantitative analysis.
Data Analysis and Expected Outcomes
This section outlines the key parameters to analyze and the typical results one can expect from a successful experiment.
Quantitative Morphological Analysis
The primary quantitative data extracted from the images are the physical dimensions of the tactoids.
Table 3: Key Quantitative Parameters for Tactoid Characterization
| Parameter | Description | Measurement Method |
|---|---|---|
| Tactoid Length | The long axis of the spindle-shaped structure. | Measured by drawing a line between the two tapered ends of the tactoid in the Rhodamine channel image. |
| Tactoid Width | The short axis, typically at the widest point of the tactoid. | Measured perpendicular to the long axis at the structure's center. |
| Intensity Profile | Fluorescence intensity as a function of position across the tactoid's width. | Plotted to show variation in density of microtubules or MAP65, often correlating with tactoid width [7]. |
| Colocalization | Spatial overlap of the Rhodamine (microtubule) and GFP (MAP65) signals. | Indicates where the crosslinking protein is bound within the microtubule bundle. |
Representative Results and Advanced Assays
- Successful Tactoid Formation: Under the microscope, fully formed tactoids will be clearly visible in both the red (Rhodamine, microtubules) and green (GFP, MAP65) channels. The images should show a high degree of overlap, confirming that MAP65 is colocalized with the microtubule bundles [7].
- Fluorescence Recovery After Photobleaching (FRAP): This protocol can be extended to study protein dynamics within the tactoids.
- For Microtubules: A FRAP experiment on the Rhodamine channel is expected to show no recovery of fluorescence after bleaching, demonstrating that the microtubules in the tactoid are static and immobile [20] [7].
- For MAP65: A FRAP experiment on the GFP channel will typically show a gradual recovery of fluorescence. This indicates that the MAP65 crosslinkers are dynamically exchanging with the surrounding solution. The recovery curve can be fitted to a rising exponential to quantify the recovery amplitude and time constant, providing insights into binding kinetics [20] [7].
Solving Common Problems and Fine-Tuning Tactoid Assembly Conditions
Within the field of cytoskeleton biophysics, the self-organization of microtubules into higher-order structures is a fundamental process, crucial for cellular functions such as cell division. One of the most intriguing structures formed in vitro is the microtubule tactoid—a spindle-shaped, liquid-crystal-like droplet that serves as a simplified model for the mitotic spindle [31]. A critical factor governing the transition between these organized states is the ionic strength of the surrounding buffer. This application note details how the precise modulation of salt concentration can drive the structural transformation of microtubule assemblies from finite, spindle-like tactoids into unbounded bundles, providing a key control parameter for researchers studying self-organization and biomimetic materials.
The underlying mechanism involves a delicate balance of electrostatic interactions. The antiparallel crosslinker MAP65 (a plant homologue of the PRC1/Ase1 family) is essential for forming tactoids, binding to and bridging adjacent microtubules [32] [31]. This crosslinker possesses charged domains, and its binding to the negatively charged microtubule surface is inherently electrostatic. Increasing the concentration of monovalent salts in the buffer screens these electrostatic interactions. Interestingly, while this screening does not completely negate MAP65 binding, it significantly disrupts the phase separation process that nucleates and grows tactoids, ultimately leading to a different organizational outcome: the formation of extensive, unbounded bundles [32].
Quantitative Data on Ionic Strength Effects
The following tables summarize key quantitative relationships between ionic strength and the properties of microtubule assemblies, providing a reference for experimental planning and data interpretation.
Table 1: Effect of Increasing Monovalent Salt Concentration on Microtubule Structures
| Salt Concentration | Observed Structure | Key Characteristics | Proposed Mechanism |
|---|---|---|---|
| Low Ionic Strength | Finite, spindle-like tactoids | Anisotropic liquid droplets; nematic order [31] | MAP65-driven phase separation is enabled [32] |
| High Ionic Strength | Unbounded length bundles | Loss of finite tactoid structure; elongated bundles | Screening of electrostatic interactions disrupts phase separation nucleation [32] |
Table 2: Impact of Ionic Strength on MAP65 Crosslinker Function
| Parameter | Low Ionic Strength | High Ionic Strength | Measurement Method |
|---|---|---|---|
| MAP65 Binding Affinity | High | Reduced, but not abolished [32] | Microtubule pelleting assays [32] |
| Inter-MAP65 Binding | Enabled | Disrupted [32] | Single-molecule binding assays [32] |
| Primary Organizational Driver | Liquid-liquid phase separation (LLPS) [32] | Direct crosslinking without LLPS [32] | Structural observation (microscopy) |
Experimental Protocols
This section provides detailed methodologies for reproducing key experiments on ionic-strength-dependent microtubule self-organization.
Protocol: Probing Ionic Strength Effects on Tactoid Formation
This protocol is adapted from established methods for self-assembling microtubule tactoids [32] [31].
I. Materials and Reagents
- Tubulin: Purified αβ-tubulin heterodimers.
- Crosslinker: MAP65 protein (or homologs PRC1/Ase1).
- GTP: Guanosine triphosphate, for microtubule polymerization.
- GMPCPP: A non-hydrolyzable GTP analog for nucleating stable, short microtubules [31].
- Buffers: BRB80 buffer (80 mM PIPES, 1 mM MgCl₂, 1 mM EGTA, pH 6.9) with varying concentrations of KCl or NaCl to adjust ionic strength.
- Crowding Agent: Polyethylene Glycol (PEG, MW ~20kDa) or dextran [31].
II. Step-by-Step Procedure
- Prepare Short Microtubule Seeds: Polymerize tubulin (e.g., at 5 mg/mL) in the presence of GMPCPP to form short, stable microtubules. This can be done by incubating the mixture at 37°C for 30-60 minutes. These seeds will serve as nucleation sites for longer filaments in the subsequent step [31].
- Grow Microtubules: Dilute the seeds into a solution containing tubulin, GTP, and an antifade system to promote dynamic growth. Incubate to achieve a population of microtubules of the desired length. For tactoid formation, shorter microtubules are often optimal [31].
- Form Tactoids at Low Ionic Strength:
- Prepare a low-salt BRB80 buffer (e.g., without added KCl).
- Mix the polymerized microtubules with MAP65 (at a molar ratio determined empirically, e.g., 1:10 crosslinker:tubulin) and a crowding agent (e.g., 1% PEG) in the low-salt buffer.
- Incubate the mixture at room temperature for 15-60 minutes and observe.
- Induce Bundle Formation at High Ionic Strength:
- Prepare a high-salt BRB80 buffer (e.g., supplemented with 100-150 mM KCl).
- Repeat the mixing and incubation steps from (3) using the high-salt buffer.
- Characterization:
- Fluorescence Microscopy: Use samples with a small fraction of fluorescently labeled tubulin or MAP5 to visualize the structures. Tactoids appear as bright, spindle-shaped droplets, while bundles are long and filamentous [32] [31].
- Fluorescence Recovery After Photobleaching (FRAP): To probe the fluidity and dynamics within the structures. Tactoids often show rapid fluorescence recovery, indicating a liquid-like interior, whereas bundles may exhibit slower or limited recovery [31].
Protocol: Measuring MAP65 Binding via Microtubule Pelleting
This assay quantitatively assesses how ionic strength affects the binding of MAP65 to microtubules [32].
I. Materials and Reagents
- MAP65 protein.
- Taxol-stabilized microtubules.
- BRB80 buffers with a range of KCl concentrations (e.g., 0, 50, 100, 150 mM).
II. Step-by-Step Procedure
- Prepare Samples: Combine a fixed concentration of MAP65 with taxol-stabilized microtubules in a series of tubes, each containing BRB80 with a different KCl concentration. Include a control with no microtubules for each salt condition.
- Incubate: Allow the binding reaction to proceed at room temperature for 20-30 minutes.
- Ultracentrifugation: Pellet the microtubules and their bound proteins using a high-speed ultracentrifuge (e.g., 100,000 x g for 20 minutes at 25°C).
- Analysis:
- Carefully separate the supernatant (unbound fraction) from the pellet (bound fraction).
- Analyze both fractions by SDS-PAGE.
- Quantify the amount of MAP65 in the pellet and supernatant bands using gel densitometry.
- Plot the fraction of MAP65 bound against the ionic strength to determine the dissociation constant (Kd) at each condition.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Microtubule Self-Assembly Studies
| Reagent / Material | Function / Explanation | Example Use Case |
|---|---|---|
| MAP65 / PRC1 | Antiparallel microtubule crosslinker; essential for driving self-organization into tactoids and bundles [32] [31]. | Core component for inducing tactoid formation in the presence of crowders. |
| GMPCPP | Non-hydrolyzable GTP analog; used to nucleate short, stable microtubule seeds that resist dynamic instability [31]. | Creating a defined population of short microtubules ideal for tactoid self-assembly. |
| PEG / Dextran | Macromolecular crowding agent; mimics the crowded cellular environment and promotes phase separation by the depletion effect [33] [31]. | Accelerating the kinetics of both supramolecular polymerization and liquid-liquid phase separation. |
| BRB80 Buffer | Standard biochemical buffer for tubulin and microtubule work, providing pH stabilization and essential cations (Mg²⁺) [31]. | The foundational aqueous environment for all in vitro microtubule experiments. |
| KCl / NaCl | Monovalent salts; used to precisely modulate the ionic strength of the buffer solution. | Experimental variable for screening electrostatic interactions and controlling assembly morphology [32]. |
Workflow and Pathway Visualization
The following diagram synthesizes the experimental and conceptual pathway through which ionic strength dictates the structural outcome of microtubule self-organization.
Managing Tubulin Stability and Preventing Denaturation During Preparation
The self-assembly of microtubule tactoids, which are spindle-shaped, liquid-crystal-like structures, provides a powerful minimal system for studying the biophysical principles underlying the formation of the mitotic spindle [20]. The successful reconstitution of these assemblies in vitro depends critically on the stability and biological activity of their fundamental building block: the tubulin heterodimer. Tubulin is a notoriously fragile protein, susceptible to denaturation, surface interactions, and proteolytic degradation, which can irrevocably compromise experimental outcomes [20] [7]. This application note details essential protocols and strategies for managing tubulin stability during preparation, specifically framed within the context of microtubule tactoid research. The procedures outlined here are integral to a broader thesis on achieving reproducible self-organization of microtubules with a minimal set of components, namely the antiparallel crosslinker MAP65 and crowding agents [20] [1]. By providing researchers with standardized, detailed methodologies for tubulin handling, we aim to enhance the reliability and reproducibility of experiments aimed at deciphering the self-organizing principles of the cellular cytoskeleton.
Fundamental Principles of Tubulin Biochemistry and Stability
Tubulin is a heterodimeric protein composed of α- and β-subunits, each with a molecular weight of approximately 50 kDa [34]. Its structural integrity and capacity for GTP-dependent polymerization into microtubules are the cornerstones of its function. Several key factors must be managed to preserve these properties from protein preparation through to experimental use.
- Cold Instability: Unlike many proteins, tubulin is cold-labile. Storage and handling on ice is essential, as exposure to room temperature can rapidly lead to denaturation and loss of function [20] [7].
- GTP Dependence: Polymerization requires GTP bound to the exchangeable site (E-site) on β-tubulin. This GTP is hydrolyzed during assembly, and the resulting GDP lattice is inherently unstable. Therefore, a fresh supply of GTP is crucial for successful polymerization experiments [35] [36].
- Surface Denaturation: Tubulin readily denatures at air-water interfaces and on certain surfaces. The use of crowding agents and surface-passivated chambers is vital to prevent this [20].
- Protease Sensitivity: As with any protein, contamination by proteases will lead to degradation. The use of pure reagents and appropriate buffer conditions is necessary to maintain protein integrity.
The conformational stability of tubulin is exemplified by studies with the drug Taxol. Hydrogen/deuterium exchange experiments have demonstrated that Taxol binding to β-tubulin induces long-range structural rigidification, markedly reducing deuterium incorporation in both β- and α-tubulin chains [35]. This indicates a pronounced increase in conformational stability that protects the polymer from depolymerization.
Essential Reagents and Buffers for Tubulin Stability
The foundation of stable tubulin begins with a properly formulated buffer. The composition of a standard General Tubulin Buffer is detailed in Table 1, and a curated list of key reagents for tactoid assembly is provided in Table 2.
Table 1: Composition of General Tubulin Buffer
| Component | Concentration | Function | Critical Notes |
|---|---|---|---|
| PIPES | 80 mM, pH 7.0 | Buffering Agent | Optimal pH range for tubulin polymerization is 6.8-7.0. |
| Magnesium Chloride (MgCl₂) | 2 mM | Cofactor | Essential for GTP binding and polymerization. |
| EGTA | 0.5 mM | Chelating Agent | Chelates calcium, a potent microtubule destabilizer. |
| GTP | 1 mM (supplement) | Nucleotide | Labile; must be added fresh just before use. Do not store [36]. |
| Glycerol | 5-10% (optional) | Stabilizer/Polymerization Enhancer | Enhances polymerization yield but is not required for maintaining biological activity [36]. |
Table 2: Research Reagent Solutions for Microtubule Tactoid Assembly
| Reagent | Function in Experiment | Key Consideration |
|---|---|---|
| Tubulin Heterodimer (unlabeled/fluorophore-labeled) | Core structural protein for microtubule polymerization. | Lyophilized powder stored at -80°C; requires careful reconstitution in cold buffer [20] [7]. |
| MAP65 (with GFP tag) | Antiparallel microtubule crosslinker; promotes self-organization into tactoids [20]. | Plant-derived homolog of PRC1/Ase1; GFP tag allows visualization of protein localization [20]. |
| GMPCPP | Non-hydrolyzable GTP analog; nucleates and stabilizes short microtubules [20]. | Creates a high density of short, stable filaments required for tactoid formation instead of long bundles [20] [37]. |
| Polyethylene Glycol (PEG) | Crowding agent; creates depletion forces that increase local tubulin concentration [20]. | Mimics macromolecular crowding in the cytoplasm, promoting condensation and assembly. |
| Pluronic F-127 | Non-ionic surfactant; prevents surface denaturation of tubulin in flow chambers [7]. | Critical for passivating glass surfaces to maintain protein functionality. |
| Glucose Oxidase/Catalase System | Oxygen-scavenging system; prevents photodamage during fluorescence imaging [7]. | Protects samples during prolonged microscopy, enabling FRAP and time-lapse imaging. |
| Dithiothreitol (DTT) | Reducing agent; prevents oxidation of cysteine residues in tubulin and other proteins. | Maintains proteins in a reduced, functional state. |
Detailed Experimental Protocol for Tubulin Preparation and Tactoid Assembly
This protocol is adapted from the established method for self-assembling microtubule tactoids [20] [7] [1]. The workflow, from coverslip preparation to imaging, is summarized in Figure 1 below.
Figure 1. Workflow for Microtubule Tactoid Self-Assembly. This diagram outlines the key stages of the protocol, highlighting the critical tubulin preparation and handling steps.
Protocol: Coverslip Silanization for Surface Passivation
Objective: To create hydrophobic coverslips that allow a block copolymer to bind and form a polymer brush, preventing microtubule and tubulin adhesion and denaturation [20].
Materials:
- Glass coverslips
- Dimethyldichlorosilane (DDS) - Highly toxic; handle in a fume hood with gloves
- Acetone (100%)
- Ethanol (100%)
- KOH (0.1 M)
- ddH₂O
- UV-Ozone (UVO) machine or plasma chamber
- Metal coverslip racks
Procedure:
- Rinse: Rinse coverslips sequentially with ddH₂O, 70% ethanol, and ddH₂O. Dry with lint-free wipes between rinses.
- UV-Ozone Clean: Place coverslips in a metal rack and irradiate in a UVO machine for 20 minutes to remove organic contaminants and background fluorescence.
- Solvent Rinse: Transfer coverslips to a dedicated silanization rack.
- Immerse in 100% acetone for 1 hour.
- Rinse container 3x with tap water and 3x with ddH₂O.
- Immerse in 100% ethanol for 10 minutes.
- Rinse container 3x with tap water and 3x with ddH₂O.
- Base Wash: Immerse rack in 0.1 M KOH for 15 minutes. Rinse container 3x with tap water and 3x with ddH₂O.
- Final Water Rinse: Immerse rack in ddH₂O three times for 5 minutes each.
- Dry: Air-dry the rack with coverslips overnight in a fume hood.
- Silanize: Immerse completely dry rack and coverslips in 2% DDS for 5 minutes.
- Post-Silanization Rinse:
- Immerse rack 2x in 100% ethanol for 5 minutes.
- Rinse container 3x with tap water and 3x with ddH₂O.
- Immerse rack 3x in ddH₂O for 5 minutes each.
- Final Dry: Air-dry the rack with coverslips overnight in a fume hood.
- Storage: Transfer silanized coverslips to boxes using tweezers. They are stable for 1-2 months.
Protocol: Tubulin Preparation and Stability Management
Objective: To properly reconstitute and store tubulin in a manner that preserves its polymerization competence [20] [7].
Materials:
- Lyophilized, unlabeled tubulin (1 mg)
- Lyophilized, rhodamine-labeled tubulin (20 µg)
- Cold PEM-80 Buffer (80 mM PIPES pH 6.9, 2 mM MgCl₂, 0.5 mM EGTA)
- Liquid nitrogen
- Ice
Procedure:
- Thaw: Remove one aliquot each of unlabeled and labeled tubulin from the -80°C freezer. Keep them on ice at all times.
- Reconstitute Unlabeled Tubulin: Add 200 µL of cold PEM-80 to the 1 mg unlabeled tubulin vial to achieve a concentration of 5 mg/mL. Keep on ice for 10 minutes to dissolve all lyophilate without agitation.
- Reconstitute Labeled Tubulin: Add 4 µL of cold PEM-80 to the 20 µg rhodamine-labeled tubulin vial. Keep on ice for 10 minutes.
- Mix: Add 100 µL of the resuspended unlabeled tubulin to the 4 µL of rhodamine-labeled tubulin. Mix by pipetting slowly 6-7 times to avoid frothing and surface denaturation.
- Aliquot and Flash-Freeze: Distribute the tubulin mix into 7 new tubes, 15 µL per tube. Immediately freeze each aliquot by immersing the tube in liquid nitrogen.
- Storage: Store all aliquots at -80°C for future use. Avoid repeated freeze-thaw cycles.
Protocol: Tactoid Assembly Experiment
Objective: To form self-organized microtubule tactoids in a surface-passivated flow chamber [20] [7].
Materials:
- Silanized coverslips and glass slides
- Double-sided tape
- Tubulin aliquot (from previous section)
- MAP65 (with GFP tag)
- GMPCPP
- Pluronic F-127 (5% in PEM-80)
- PEG (crowding agent)
- Glucose oxidase/catalase mix
- Dithiothreitol (DTT)
- PEM-80 buffer
- 5-minute epoxy
Procedure:
- Construct Flow Chamber:
- Clean a glass slide with ddH₂O and ethanol.
- Create two thin strips from double-sided tape and place them 5-8 mm apart on the slide.
- Place a silanized coverslip on top of the tape strips to create a flow path. Press gently to seal.
- Coat Chamber: Introduce 20 µL of 5% Pluronic F-127 into the flow chamber. Place the chamber in a humidified Petri dish for 5-7 minutes. This coats the surface and prevents tubulin denaturation.
- Prepare Reaction Mix (on ice): In a sterile tube on ice, combine the following components by gentle pipetting:
- 5 µL PEM-80
- 1 µL GMPCPP (to nucleate short, stable microtubules)
- 1 µL Pluronic F-127
- 1 µL DTT
- 1 µL Glucose
- 4 µL Polyethylene Glycol (PEG)
- 2.5 µL of the prepared tubulin mix
- 1.5 µL MAP65 (with GFP for visualization)
- Add Enzymes and Load: Add 1 µL of the pre-mixed glucose oxidase/catalase solution to the tube and mix by pipetting 7-8 times. Remove the Pluronic solution from the flow chamber by wicking with a lint-free wipe and immediately add the tubulin-MAP65 reaction mix to the chamber.
- Seal and Incubate: Seal both ends of the chamber with 5-minute epoxy. Transfer the chamber to an environmental chamber set to 37°C for 30 minutes to allow for microtubule nucleation and tactoid growth.
- Image: After incubation, image the tactoids using a fluorescence microscope with a 60x or higher magnification objective (NA ≥ 1.2). Use a 561 nm laser for rhodamine-tubulin and a 488 nm laser for GFP-MAP65. Acquire at least 10 images from different areas to capture over 100 tactoids for statistical analysis.
Troubleshooting and Quality Control
A successful tactoid experiment results in the formation of numerous spindle-shaped assemblies visible in both the tubulin and MAP65 channels, with fluorescence intensities that perfectly overlap [7]. Common issues and their solutions are:
- No Tactoids Formed: This is often due to degraded tubulin. Ensure tubulin is handled on ice, flash-frozen properly, and has not been subjected to multiple freeze-thaw cycles. Verify that the GTP/GMPCPP is fresh.
- Large, Irregular Bundles Instead of Tactoids: This can occur if microtubules are too long. Ensure GMPCPP is used to generate short, stable seeds and that the concentration of MAP65 is optimized [20].
- Poor Fluorescence or High Background: Check the activity of the oxygen-scavenging system. Ensure the flow chambers are properly sealed during incubation to prevent evaporation.
- Tubulin Aggregation: This indicates surface denaturation or contamination. Ensure all surfaces are properly passivated with Pluronic F-127 and that all buffers are prepared with high-purity reagents.
Meticulous management of tubulin stability is not merely a preparatory step but a foundational requirement for the successful self-assembly of microtubule tactoids. The protocols detailed herein—from the precise formulation of buffers and careful reconstitution of tubulin to the strategic passivation of surfaces—are designed to shield this labile protein from the myriad factors that compromise its function. By integrating these practices, researchers can achieve the highly reproducible, spindle-like microtubule assemblies necessary to probe the minimal physical and biochemical conditions governing cytoskeletal organization. This reliability opens the door for future investigations using diverse crosslinking proteins, motor proteins, and stabilizing drugs to further deconstruct the remarkable self-organization of the mitotic spindle.
Microtubule tactoids are spindle-like, self-organized assemblies that serve as valuable in vitro models for studying the mitotic spindle and liquid crystal behavior of the cytoskeleton. Achieving consistent nucleation of these structures is a common challenge, primarily governed by the precise interplay of tubulin concentration, crosslinking proteins, and reaction timing. This application note provides a systematic troubleshooting guide, grounded in established protocols, to diagnose and resolve poor tactoid nucleation. The guidance is framed within the context of advanced research on cytoskeletal self-assembly, providing drug development professionals and scientists with the quantitative data and methodologies necessary to robustly reconstitute these complex biomolecular assemblies.
The following tables consolidate the key quantitative factors influencing tactoid nucleation. Optimizing these parameters is essential for successful assembly.
Table 1: Critical Reagent Concentrations for Tactoid Assembly
| Parameter | Optimal Concentration Range | Effect of Low Concentration | Effect of High Concentration |
|---|---|---|---|
| αβ-tubulin | 7 µM and above [38] | No nucleation observed below critical concentration [38] | Increased nucleation rate; potential for uncontrolled aggregation [2] |
| MAP65 | Not explicitly quantified (see protocol) | Insufficient crosslinking; no stable assemblies [2] | Over-bundling; large, amorphous aggregates [2] |
| Crowding Agent (PEG) | Not explicitly quantified (see protocol) | Insufficient depletion force to condense microtubules [2] | Excessive condensation, destabilizing tactoid shape [2] |
| GMPCPP | Used for nucleation/stabilization [2] | Microtubules too long/dynamic, forming long bundles instead of tactoids [2] | N/A |
Table 2: Kinetic and Cooperativity Factors in Nucleation
| Factor | Value / Characteristic | Experimental Implication |
|---|---|---|
| γ-TuRC Nucleation Cooperativity | 3.9 ± 0.5 αβ-tubulin dimers [38] | The rate-limiting step is the cooperative assembly of ~4 tubulin dimers on the nucleator. |
| Spontaneous Nucleation Cooperativity | 8.1 ± 0.9 αβ-tubulin dimers [38] | Spontaneous nucleation requires a much higher energetic barrier and tubulin concentration (~14 µM) [38]. |
| Microtubule Length | Short, stabilized filaments (via GMPCPP) [2] | Long microtubules form bundles; short filaments are essential for tapered tactoid formation [2]. |
Core Experimental Protocol for Tactoid Assembly
This section details the foundational protocol for forming microtubule tactoids, with integrated troubleshooting notes.
Materials and Reagent Setup
- Tubulin Preparation: Purified αβ-tubulin dimers. For fluorescence microscopy, a fraction should be labeled with a compatible fluorophore (e.g., Alexa Fluor 488). Prior to assembly, tubulin should be aliquoted and stored at -80°C to prevent degradation [39].
- MAP65 Purification: Express and purify the plant-based, antiparallel microtubule crosslinker MAP65 (a homolog of PRC1 and Ase1) [2] [1] [6]. This protein is central to inducing the self-organization of microtubules into spindle-like assemblies.
- Assembly Buffer: A BRB80-like buffer (80 mM PIPES, 1 mM MgCl₂, 1 mM EGTA, pH 6.8) is standard. Supplement with 1 mM GTP and an ATP-regenerating system for motor proteins if used.
- Crowding Agent: Prepare a stock solution of Polyethylene Glycol (PEG) at a molecular weight and concentration determined empirically (e.g., 20-40 kDa). PEG creates depletion forces that condense microtubules [2].
- Stabilizing Nucleotide: Use GMPCPP, a non-hydrolyzable GTP analog, to nucleate and stabilize short microtubule seeds. This prevents dynamic instability and end-to-end annealing, which is crucial for forming tactoids instead of long bundles [2].
- Silanized Coverslips: Prepare glass coverslips treated with dimethyldichlorosilane (DDS) to create a hydrophobic surface for subsequent polymer brush coating, preventing non-specific microtubule adhesion [2].
- Safety Note: DDS is highly toxic. This step must be performed in a fume hood while wearing appropriate personal protective equipment [2].
Step-by-Step Assembly Procedure
Prepare Short Microtubule Seeds:
- Mix αβ-tubulin (final concentration ≥7 µM) with GMPCPP in assembly buffer.
- Incubate at 37°C for 30-60 minutes to polymerize short, stabilized microtubules.
- Troubleshooting Tip: The length distribution of seeds is critical. If tactoids do not form, analyze seed length via electron microscopy or TIRF and optimize incubation time [2].
Form the Tactoid Assembly Mix:
- Combine the following components in a tube on ice:
- Short microtubule seeds (from Step 1)
- Purified MAP65 protein
- Crowding agent (PEG)
- Additional assembly buffer to achieve final desired concentrations.
- Mix the solution gently by pipetting. Avoid introducing air bubbles.
- Combine the following components in a tube on ice:
Incubate for Tactoid Formation:
- Transfer the mixture to a room temperature (20-25°C) environment.
- Allow the reaction to proceed for 30-90 minutes.
- Troubleshooting Tip: If nucleation is slow or yields are low, systematically vary the incubation time and the ratio of MAP65 to microtubules. A time-course experiment can identify the optimal nucleation window [2].
Image and Characterize:
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Microtubule Tactoid Research
| Reagent | Function in Tactoid Assembly | Key Characteristics |
|---|---|---|
| MAP65 / Ase1 / PRC1 | Antiparallel microtubule crosslinker [2] [1] [6] | Self-organizes microtubules into spindle-like structures; dimeric structure with self-binding affinity [2]. |
| GMPCPP | Microtubule stabilizing nucleotide [2] | Generates short, stable microtubule seeds; prevents dynamic instability and annealing. |
| PEG (Polyethylene Glycol) | Crowding agent [2] | Induces depletion forces that condense microtubules into higher-order assemblies. |
| XMAP215 | Microtubule polymerase & nucleation factor [40] [41] | Functions synergistically with γ-TuRC to drastically increase microtubule nucleation efficiency; not part of core tactoid protocol but key nucleation agent [38]. |
| γ-TuRC (γ-Tubulin Ring Complex) | Core microtubule nucleator [39] [38] | Templates microtubule growth; requires cooperative binding of ~4 tubulin dimers for nucleation [38]. |
Workflow and Pathway Diagrams
The following diagrams outline the logical relationship between key parameters and the experimental workflow for troubleshooting.
Diagram Title: Troubleshooting Logic for Poor Tactoid Nucleation
Diagram Title: Core Experimental Workflow for Tactoid Assembly
Adjusting Crosslinker and Crowder Concentrations for Desired Assembly Morphology
Within the cellular milieu, the cytoskeleton undergoes remarkable self-organization without centralized control, particularly during cell division when microtubules form the mitotic spindle. This structure shares striking similarities with liquid crystal tactoids—spindle-shaped, anisotropic droplets that nucleate and grow from an isotropic phase [12]. Recent advances have demonstrated that reconstituting these microtubule tactoids in vitro is achievable with a minimal system comprising microtubules, specific crosslinking proteins, and crowding agents [12] [1]. This application note provides detailed protocols and optimization strategies for controlling the morphology of these assemblies by precisely adjusting the concentrations of crosslinkers and crowders, enabling researchers to create in vitro models that mimic biological structures and inform drug development efforts.
The Science of Microtubule Tactoids
Fundamental Concepts
Microtubule tactoids are mesoscale assemblies where microtubules act as mesogens (the fundamental units of liquid crystals) [12] [6]. Their formation represents an exciting intersection of cell biology and soft matter physics, where biological filaments self-organize through physical principles including entropy-driven phase separation [33]. These structures are "solid-like condensates" that maintain a limited width while extending in length, differing from traditional liquid crystal organization [42].
The shape and internal order of tactoids emerge from a delicate balance between several energy contributions: the bending stiffness of individual microtubules, the interaction energies provided by crosslinkers, and the entropic forces induced by macromolecular crowders [42] [43]. Understanding these interactions is crucial for controlling assembly morphology for specific research applications.
Key Components for Self-Assembly
Successful reconstitution requires three essential components:
- Microtubules: Short, stabilized microtubules are crucial, typically achieved using GMPCPP, a non-hydrolyzable GTP analog that prevents dynamic instability and promotes uniformity [12].
- Crosslinkers: MAP65 (and its homologs Ase1 in yeast and PRC1 in mammals) serves as an antiparallel crosslinker that specifically organizes microtubules into bipolar, spindle-like assemblies [12] [1] [6].
- Crowding Agents: Polyethylene glycol (PEG) or dextran create volume exclusion effects that enhance local tubulin concentration and promote phase separation into tactoids [12] [42] [33].
Table 1: Essential Research Reagents for Microtubule Tactoid Assembly
| Reagent | Function | Key Variants | Considerations |
|---|---|---|---|
| Tubulin | Structural polymer | Purified from tissue or recombinant sources; fluorescently-labeled variants available for visualization | Use GMPCPP to create short, stable seeds; typical working concentrations 5-20 μM |
| MAP65 | Antiparallel crosslinking | Plant MAP65, yeast Ase1, mammalian PRC1 | Concentration directly controls bundle density and tactoid dimensions [12] |
| PEG | Crowding agent | Various molecular weights (e.g., 8-40 kDa) | Higher molecular weight and concentration increase depletion forces [42] |
| Dextran | Alternative crowder | MW ≈ 500 kDa commonly used | Affects both kinetics and final morphology; modulates fluidity of condensates [42] [33] |
| GMPCPP | Microtubule stabilizer | Non-hydrolyzable GTP analog | Creates short, stable microtubule seeds resistant to dynamic instability [12] |
Optimization Strategies for Assembly Morphology
Crosslinker Concentration Effects
The MAP65 crosslinker concentration fundamentally determines the structural integrity and dimensional control of resulting tactoids. As an antiparallel crosslinker, MAP65 preferentially binds overlapping microtubules in specific orientations, promoting the formation of bipolar assemblies reminiscent of mitotic spindles [12] [6].
Optimization Guidelines:
- Low concentrations (10-50 nM) produce loose, irregular assemblies with limited anisotropy
- Intermediate concentrations (50-200 nM) generate well-defined tactoids with high aspect ratios
- High concentrations (>200 nM) may create overly dense bundles lacking the tapered ends characteristic of tactoids
The exact optimal concentration depends on microtubule density and length, requiring empirical determination for each preparation. Fluorescence recovery after photobleaching (FRAP) experiments demonstrate that higher MAP65 concentrations significantly reduce component mobility within tactoids, indicating a transition toward more solid-like material properties [12].
Crowder Influence on Tactoid Formation
Crowding agents like PEG and dextran operate through entropic volume exclusion - they occupy space in the solution, effectively increasing the local concentration of microtubules and crosslinkers while reducing the entropic penalty for alignment [42] [33].
Dextran Optimization:
- Low dextran (0-0.5 wt%): Slow phase separation, large tactoid formation
- Intermediate dextran (0.5-1.5 wt%): Rapid formation of high-aspect-ratio tactoids
- High dextran (>3 wt%): Filamentous bundles rather than distinct tactoids
Recent research has revealed a bell-shaped curve for aspect ratio as a function of dextran concentration, with a maximum at approximately 0.9 wt% [33]. Higher dextran concentrations not only accelerate the kinetics of tactoid formation but also produce smaller, more anisotropic droplets with enhanced internal order [42] [33].
Table 2: Crosslinker and Crowder Optimization Guide
| Parameter | Low Concentration | Medium Concentration | High Concentration |
|---|---|---|---|
| MAP65 (nM) | 10-50 | 50-200 | >200 |
| Resulting Morphology | Loose, irregular assemblies | Well-defined, high-aspect-ratio tactoids | Overly dense bundles, reduced anisotropy |
| PEG/Dextran | 0-2% | 2-5% | >5% |
| Resulting Morphology | Large, sparse tactoids | Uniform, spindle-shaped tactoids | Small, highly ordered assemblies approaching solid state |
| Aspect Ratio | Low (2-4) | High (5-10) | Variable, often lower |
| Formation Kinetics | Slow (hours) | Moderate (30-60 min) | Fast (<30 min) |
Experimental Protocols
Microtubule Tactoid Assembly
This protocol describes the core methodology for reconstituting microtubule tactoids in vitro, adapted from established visualized experiments [12] [1].
Materials:
- Purified tubulin (≥95% pure)
- MAP65 (or homolog crosslinker)
- PEG (20 kDa) or dextran (500 kDa)
- GMPCPP
- BRB80 buffer (80 mM PIPES, 1 mM MgCl₂, 1 mM EGTA, pH 6.8)
- Silanized coverslips
- Flow chambers
Procedure:
- Prepare short microtubule seeds:
- Mix tubulin (5-10 μM) with 1 mM GMPCPP in BRB80 buffer
- Incubate at 37°C for 30-60 minutes
- Pellet seeds by centrifugation (100,000 × g, 10 minutes)
- Resuspend in fresh BRB80 buffer
Silanize coverslips (critical for proper surface passivation):
- Clean coverslips with ddH₂O, 70% ethanol, and ddH₂O sequentially
- Dry with lint-free wipes between washes
- Perform silanization in a fume hood using dimethyldichlorosilane (CAUTION: highly toxic)
- Cure silanized coverslips overnight
Set up flow chamber:
- Create a parallel flow chamber using silanized coverslips
- Passivate with pluronic F-127 to prevent nonspecific adhesion
Form tactoid assemblies:
- Combine in order:
- BRB80 buffer (to final volume)
- Short microtubule seeds (final concentration 1-2 μM)
- PEG or dextran (final concentration 2-5%)
- MAP65 (final concentration 50-200 nM)
- Introduce reaction mixture into flow chamber
- Seal chambers to prevent evaporation
- Combine in order:
Incubate and image:
- Maintain at room temperature (22-25°C) for 30-90 minutes
- Monitor formation by fluorescence microscopy
Characterization Methods
Fluorescence Microscopy:
- Include 1-5% fluorescently-labeled tubulin in the assembly mixture
- Image using TIRF or confocal microscopy to resolve internal structure
- Capture time-lapse sequences to monitor assembly kinetics
Fluorescence Recovery After Photobleaching (FRAP):
- Photobleach a region of interest within tactoids
- Monitor recovery over 5-30 minutes
- Calculate recovery half-time and mobile fraction to quantify material properties
Quantitative Morphometry:
- Measure tactoid length and width from microscopy images
- Calculate aspect ratio (length/width)
- Quantify alignment order parameters using Fourier analysis
Troubleshooting and Data Interpretation
Common Challenges
Irregular Assemblies:
- Cause: Overly long microtubules or incorrect crosslinker ratio
- Solution: Optimize GMPCPP concentration to ensure short seeds; titrate MAP65
Limited Tactoid Formation:
- Cause: Insufficient crowding or suboptimal surface passivation
- Solution: Increase PEG/dextran concentration (2-5% range); verify silanization
Excessive Bundling:
- Cause: Too high crosslinker concentration
- Solution: Reduce MAP65 concentration; include intermediate dilution steps
Data Interpretation Guidelines
The aspect ratio of tactoids serves as a key indicator of internal organization and material properties. Higher aspect ratios (≥5) typically indicate well-ordered nematic arrangements, while lower values suggest disordered or isotropic organization. Importantly, tactoids display limited width expansion regardless of crowder concentration, behaving as "solid-like condensates" rather than traditional liquid crystals [42].
FRAP measurements provide insight into the material state: rapid recovery indicates liquid-like properties, while limited recovery suggests solid-like behavior. MAP65 concentration strongly influences this property, with higher concentrations leading to more restricted component mobility [12].
Applications in Drug Development
The reconstituted tactoid system provides a minimal model for screening compounds that target microtubule-associated proteins or microtubule organization. Specifically:
- Anti-mitotic drug evaluation: Taxanes, vinca alkaloids, and novel compounds can be tested for their effects on tactoid stability and organization [44]
- Crosslinker-targeted therapeutics: Compounds interfering with PRC1 (the mammalian MAP65 homolog) can be screened using this minimal system
- Biomolecular condensate research: Tactoids represent a model for studying phase separation in biological systems with direct relevance to cellular organization [33]
The system's simplicity enables high-resolution imaging and quantitative analysis not easily achievable in cellular environments, while maintaining physiological relevance through the use of authentic cytoskeletal components.
The self-assembly of microtubule tactoids represents a powerful intersection of cell biology and soft matter physics, providing a minimal system to dissect the physical principles underlying cellular organization. Precision adjustment of crosslinker and crowder concentrations enables controlled manipulation of assembly morphology, yielding structures that closely mimic biological counterparts. The protocols outlined herein provide researchers with a robust foundation for exploring microtubule-based condensates, with significant potential for illuminating fundamental biological processes and accelerating drug discovery efforts targeting the cytoskeleton.
Characterizing and Validating Tactoid Structure and Component Dynamics
Within the context of a broader thesis on microtubule self-assembly, the quantitative analysis of the resulting structures is paramount. This application note provides a detailed protocol for characterizing the physical dimensions and fluorescence intensity profiles of microtubule tactoids. These spindle-shaped, liquid-crystal-like assemblies are reconstituted in vitro using a minimal set of components and serve as a model for biological self-organization, particularly in the study of the mitotic spindle [45] [6]. The procedures outlined herein are designed for researchers and scientists in cell biology and biophysics, providing robust methods to quantify self-organization phenomena.
Experimental Protocols for Tactoid Assembly and Imaging
The foundation of reliable quantitative analysis is a reproducible preparation of microtubule tactoids. The following protocols detail the assembly process and the subsequent steps for fluorescence imaging.
Protocol for Microtubule Tactoid Assembly
This protocol is adapted from established methods for reconstituting spindle-like assemblies [45] [6].
Key Reagents:
- Stable Microtubule Seeds: Nucleated and stabilized using GMPCPP to create a high density of short microtubules, which is critical for tactoid formation [45].
- Crosslinking Protein: MAP65, an antiparallel microtubule crosslinker from plants (a homolog of Ase1/PRC1), is essential for self-organization into tactoids [45] [6].
- Crowding Agent: Polyethylene glycol (PEG) is used to create depletion interactions that crowd the microtubules, promoting bundle formation [45].
Procedure:
- Prepare Microtubules: Polymerize tubulin in the presence of GMPCPP to create stable, short microtubule seeds. This step suppresses dynamic instability and allows for a high local concentration of rigid polymers [45].
- Mix Assembly Components: Combine the stabilized microtubules with the crosslinker MAP65 and the crowding agent PEG in an appropriate biochemical buffer.
- Incubate for Self-Assembly: Allow the mixture to incubate at room temperature for a specified period to enable the self-organization of microtubules into tactoids [45] [6].
Protocol for Fluorescence Microscopy
Key Reagents:
- Fluorescent Label: Use fluorophore-conjugated tubulin (e.g., HiLyte 488-tubulin) to incorporate fluorescence into the microtubule polymers for visualization.
Procedure:
- Prepare Sample Chamber: Assemble a flow cell or use a chambered coverslip for imaging.
- Immobilize Tactoids: Introduce the tactoid assembly into the chamber and allow it to settle.
- Image Acquisition: Use a high-resolution fluorescence microscope equipped with a high-sensitivity camera (e.g., an EMCCD or sCMOS camera). Acquire z-stacks of images to fully capture the three-dimensional structure of the tactoids.
Quantitative Analysis of Tactoid Morphology
This section details the methodologies for extracting quantitative data on tactoid shape and internal structure from fluorescence microscopy images.
Workflow for Image Analysis
The process of analyzing acquired images to extract quantitative data can be broken down into a series of defined steps, as visualized in the following workflow.
Measuring Dimensions and Intensity Profiles
Tactoid Dimension Analysis:
- Length and Width: After image segmentation and binarization, the major axis of the best-fitting ellipse defines the tactoid length. The minor axis, typically measured at the tactoid's midpoint, defines its width [45].
- Aspect Ratio: Calculate the aspect ratio as Length / Width. This is a key parameter describing the slenderness of the tactoid, with higher values indicating more spindle-like shapes [45].
Fluorescence Intensity Profile Analysis:
- Axial Profile: Plot the fluorescence intensity along the central, long axis of the tactoid. This profile can reveal asymmetries or gradients in microtubule density from one end of the tactoid to the other.
- Transverse Profile: Plot the fluorescence intensity along a line perpendicular to the long axis, usually at the center. The full width at half maximum (FWHM) of this profile provides a measure of the tactoid's diameter at its widest point.
Advanced Kinetic and Structural Analysis
For high-resolution analysis of microtubule dynamics within assemblies, tools like TipTracker can be employed. This semi-automated image processing tool allows for high spatial (~10-40 nm) and temporal (1-10 Hz) resolution measurements of microtubule tip position and dynamics, and can even estimate tip structure from fluorescence distribution decay [46]. While originally designed for single microtubules, its principles are applicable to the ends of microtubule bundles within tactoids.
The Scientist's Toolkit: Essential Research Reagents
The following table details key materials and their functions for experiments involving microtubule tactoids.
Table 1: Key Research Reagent Solutions for Microtubule Tactoid Assays
| Item | Function in the Experiment | Key Characteristics |
|---|---|---|
| Tubulin (with fluorescent conjugates) | The primary structural protein subunit that polymerizes to form microtubules. | High purity; conjugates like HiLyte 488-tubulin allow fluorescence visualization and quantification [47]. |
| MAP65 / Ase1 / PRC1 | An antiparallel microtubule crosslinking protein. | Essential for self-organization of short microtubules into bipolar, spindle-shaped tactoids [45] [6]. |
| GMPCPP | A non-hydrolyzable GTP analog used to nucleate and stabilize microtubules. | Creates short, stable microtubule "seeds" that resist dynamic instability, crucial for tactoid formation [45]. |
| PEG (Polyethylene Glycol) | A crowding agent that creates depletion forces. | Increases effective microtubule concentration by excluding volume, promoting bundle and tactoid assembly [45]. |
| SiR-Tubulin | A cell-permeable fluorogenic dye that labels microtubules in live cells. | Useful for live-cell imaging of microtubule dynamics with low background [47]. |
| TipTracker Software | A semi-automated image analysis tool. | Enables high-resolution tracking of microtubule tip position and estimation of tip structure [46]. |
Data Presentation and Analysis
This section summarizes the quantitative parameters that can be expected from a successful tactoid experiment and provides a framework for their analysis.
Table 2: Summary of Key Quantitative Parameters for Tactoid Analysis
| Parameter | Description | Measurement Technique | Biological/Physical Significance |
|---|---|---|---|
| Tactoid Length | The end-to-end distance along the major axis. | Fluorescence microscopy image analysis. | Indicates the scale of self-organization and the processivity of crosslinking. |
| Tactoid Width | The diameter at the midpoint, perpendicular to the long axis. | Fluorescence microscopy image analysis. | Relates to the number of microtubules bundled within the assembly. |
| Aspect Ratio | The ratio of Length to Width. | Calculated from length and width measurements. | Describes the shape, with high values indicating spindle-like, nematic tactoids [45]. |
| Intensity FWHM | The full width at half maximum of the transverse intensity profile. | Fluorescence intensity profile analysis. | A more precise measure of bundle thickness, accounting for signal distribution. |
| Tip Tracking Data | Kinetics (growth/shrinkage speeds) of microtubule ends. | High-resolution tools like TipTracker [46]. | Reveals the dynamic behavior of microtubules within the constrained tactoid environment. |
The relationships between these measured parameters form the core of the analysis, as they reveal the underlying physics of the system. The following diagram illustrates the logical pathway from raw measurements to scientific insight.
Probing Material Properties with Fluorescence Recovery After Photobleaching (FRAP)
Fluorescence Recovery After Photobleaching (FRAP) is a powerful fluorescence microscopy technique used to quantify the dynamic properties of molecules within soft materials and biological systems. By measuring the diffusion and binding interactions of fluorescently-labeled molecules, FRAP provides critical insights into material microstructure and mobility. Within the context of microtubule tactoids—spindle-like liquid crystalline assemblies formed by the self-organization of microtubules and cross-linking proteins like MAP65—FRAP serves as an essential tool for characterizing the material state and transport properties [31] [6]. This protocol details the application of FRAP to probe the material properties of microtubule tactoids, providing researchers and drug development professionals with a method to quantify molecular mobility and binding in these biologically-inspired assemblies.
Theoretical Foundation of FRAP
The quantitative analysis of FRAP recovery curves enables the determination of key kinetic parameters, primarily diffusion coefficients and binding constants. The appropriate model for analysis depends on whether the fluorescence recovery is limited by diffusion or by reaction (binding/unbinding) kinetics [48].
Diffusion-Limited Recovery
For a circular bleach spot of radius ( w ), and assuming instantaneous bleaching and a uniform initial bleached profile, the fluorescence recovery as a function of time, ( f(t) ), is given by the Soumpasis equation: [ f(t) = e^{-2\tauD / t} \left[ I0(2\tauD / t) + I1(2\tauD / t) \right] ] where ( \tauD ) is the characteristic diffusion time, and ( I0 ) and ( I1 ) are modified Bessel functions of the first kind. The diffusion coefficient ( D ) is then calculated as: [ D = \frac{w^2}{4\tau_D} ] This model applies when the recovery rate is governed by the time it takes for unbleached molecules to diffuse into the bleached region [48].
Reaction-Limited Recovery
When recovery is limited by the dissociation rate of molecules bound to static sites within the bleached area, the fluorescence recovery follows a simple exponential: [ f(t) = 1 - e^{-k{\text{off}}t ] where ( k{\text{off}} ) is the dissociation rate constant. This model is valid only when the exchange due to binding is much slower than diffusion (( 1/k_{\text{off}} \gg r^2/D )) [48].
Complex Dynamics
In heterogeneous systems like microtubule tactoids, recovery often involves both diffusion and binding interactions. Complex models incorporating both processes may be required, though parameter estimation can be challenging and may require complementary experiments, such as bleaching areas of different sizes [48].
Experimental Protocol for FRAP on Microtubule Tactoids
Reagent and Material Preparation
- Microtubule Tactoid Assembly: Follow established protocols to assemble microtubule tactoids [31] [6]. Key components include:
- Stabilized Microtubules: Short, GMPCPP-stabilized microtubules to serve as high-aspect-ratio mesogens.
- Cross-linker: MAP65 protein, an antiparallel microtubule cross-linker from plants, for promoting self-organization into spindle-like tactoids.
- Crowding Agent: Polyethylene glycol (PEG) to induce depletion interactions and promote tactoid formation.
- Fluorescent Labeling: Microtubules should be fluorescently labeled for detection. Common methods include incorporating rhodamine-labeled tubulin or using GFP fusion proteins if studying MAP65 dynamics specifically [31] [49].
Sample Preparation for Imaging
- Sample Chamber: Assemble microtubule tactoids in an imaging-compatible chamber, such as a glass-bottom dish (#1.5) [49].
- Immobilization: Ensure tactoids are immobilized for stable imaging. This can be achieved through chamber surface treatment or by working within a sufficiently viscous or gelled medium.
- Imaging Medium: Prior to image acquisition, replace the assembly medium with an appropriate imaging medium to reduce background fluorescence and maintain sample health during experimentation [49].
FRAP Data Acquisition
The following steps are performed on a confocal laser scanning microscope equipped with acousto-optic tunable filters (AOTF) for selective bleaching and a heated chamber maintained at 37°C [49].
- Initial Imaging: Save a background image of the sample before photobleaching.
- Bleaching: Focus a high-intensity laser pulse onto a small, defined region of interest (ROI) within a single microtubule tactoid. The laser wavelength should be appropriate for the fluorophore (e.g., 488 nm for GFP).
- Recovery Imaging: Immediately following the bleach pulse, acquire a time-lapse sequence of images of the bleached area at low laser intensity to monitor the fluorescence recovery over time. The acquisition frequency should be optimized for the expected timescale of recovery.
Data Analysis
- Background Correction: Subtract background fluorescence from all images.
- Fluorescence Quantification: Measure the mean fluorescence intensity within the bleached ROI, a reference unbleached area within the tactoid, and a background area for each time point.
- Normalization: Normalize the bleach spot intensity to correct for general photobleaching during acquisition, typically by dividing by the intensity in the unbleached reference area.
- Curve Fitting: Fit the normalized recovery curve to the appropriate mathematical model (e.g., Soumpasis for diffusion-limited recovery) to extract the characteristic recovery time (( \tauD )) or dissociation rate (( k{\text{off}} )).
- Parameter Calculation: Calculate the diffusion coefficient ( D ) or other relevant kinetic parameters from the fitted values.
Quantitative Parameters in FRAP Analysis
The table below summarizes the core quantitative parameters derived from FRAP experiments.
Table 1: Key Quantitative Parameters from FRAP Analysis
| Parameter | Symbol | Unit | Description |
|---|---|---|---|
| Characteristic Diffusion Time | ( \tau_D ) | s | The time for half-maximal recovery of fluorescence in a diffusion-limited process. |
| Diffusion Coefficient | ( D ) | µm²/s | A measure of the rate of molecular motion through the medium. |
| Mobile Fraction | ( M_f ) | % | The proportion of molecules that are free to diffuse into the bleached area. |
| Immobile Fraction | ( I_f ) | % | The proportion of molecules that are permanently or transiently bound and do not contribute to recovery. |
| Dissociation Rate Constant | ( k_{\text{off}} ) | s⁻¹ | The rate at which bound molecules dissociate from their binding sites. |
Essential Research Reagent Solutions
The following table details key reagents and materials required for FRAP analysis of microtubule tactoids.
Table 2: Essential Research Reagents and Materials for FRAP on Microtubule Tactoids
| Item | Function/Application in Protocol | Key Considerations |
|---|---|---|
| Tubulin (Rhodamine-labeled) | Core structural protein of microtubules; fluorescent label enables visualization and photobleaching. | Use GMPCPP to nucleate and stabilize short filaments [31]. |
| MAP65 Protein | Antiparallel microtubule cross-linker; drives self-organization into spindle-like tactoids [31] [6]. | A homolog of mammalian PRC1; specific cross-linking is essential for tactoid shape. |
| Polyethylene Glycol (PEG) | Crowding agent; induces depletion interactions that promote microtubule condensation and tactoid formation [31]. | Molecular weight and concentration affect the strength of depletion forces. |
| Glass Bottom Dishes | Imaging chamber; provides optimal optical clarity for high-resolution fluorescence microscopy. | #1.5 thickness is ideal for most high-NA oil immersion objectives [49]. |
| Confocal Microscope with AOTF | Instrumentation for FRAP; allows precise selection of ROI for bleaching and controlled low-intensity imaging for recovery. | A heated stage (37°C) is often necessary to maintain protein activity. |
Workflow and Analytical Pathway
The following diagram illustrates the complete experimental and analytical workflow for a FRAP experiment on microtubule tactoids.
FRAP Experimental and Analytical Workflow
This application note provides a detailed protocol for applying FRAP to characterize the material properties of microtubule tactoids. By following the outlined procedures for sample preparation, data acquisition, and quantitative analysis, researchers can extract precise kinetic parameters that define the dynamic environment within these self-assembled structures. The ability to probe mobility and binding in a minimal in vitro system like microtubule tactoids is invaluable for understanding the physical principles underlying more complex cellular structures, such as the mitotic spindle, and for informing the design of biomimetic materials in drug development and synthetic biology.
Fluorescence Recovery After Photobleaching (FRAP) serves as a powerful quantitative technique for analyzing the dynamic properties of molecules within cellular structures and in vitro reconstitutions. This Application Note details the protocol for and interpretation of FRAP experiments conducted on microtubule tactoids—minimalistic, spindle-like assemblies formed through self-organization. The core finding is a striking dichotomy: while the microtubule lattice within these tactoids is immobile, the Microtubule-Associated Protein 65 (MAP65) that crosslinks them exhibits significant mobility. This contrast provides key insights into the stability and maintenance of such biomolecular condensates, with direct implications for understanding the mitotic spindle machinery [1] [2] [7].
The mitotic spindle, a microtubule-based structure essential for chromosome segregation, exhibits properties of a liquid crystal tactoid—a nematic phase droplet nucleated from an isotropic fluid. Reconstituting spindle-like structures in vitro provides a reductionist approach to deciphering the physical principles of its self-organization. A key protocol involves using short, stabilized microtubules and the plant antiparallel microtubule crosslinker MAP65 (a homolog of mammalian PRC1 and yeast Ase1) to form stable, spindle-shaped bundles known as microtubule tactoids [2] [6].
To probe the material properties and molecular dynamics within these assemblies, researchers employ Fluorescence Recovery After Photobleaching (FRAP). In a FRAP experiment, a region of interest within a fluorescently labeled structure is photobleached with a high-intensity laser, and the subsequent recovery of fluorescence into this region is monitored over time. The recovery kinetics reveal the mobility and binding kinetics of the labeled molecules [50].
This note integrates the tactoid self-assembly protocol with a FRAP assay, specifically highlighting how to interpret the divergent behaviors of the microtubule scaffold and its associated crosslinker.
The Scientist's Toolkit: Essential Reagents for Tactoid Assembly and FRAP
Table 1: Key reagents and their functions in the microtubule tactoid assembly and FRAP protocol.
| Reagent | Function/Description |
|---|---|
| Tubulin | Structural protein subunit of microtubules; unlabeled and fluorescently-labeled (e.g., Rhodamine) versions are mixed for visualization [7]. |
| MAP65 | Plant-derived, antiparallel microtubule crosslinker; can be fused to GFP for visualization. Essential for tactoid formation [2] [22]. |
| GMPCPP | A non-hydrolyzable GTP analog used to nucleate and stabilize short microtubules, preventing dynamic instability and annealing [2]. |
| PEG (Polyethylene Glycol) | A crowding agent that creates depletion forces, increasing the effective concentration of microtubules and promoting bundle condensation [2]. |
| Glucose Oxidase/Catalase | An oxygen-scavenging system to minimize fluorophore photobleaching during imaging [7]. |
| Silanized Coverslips | Coverslips treated with dimethyldichlorosilane to create a hydrophobic surface for subsequent polymer brush coating, preventing nonspecific microtubule adhesion [2]. |
Experimental Protocol
Self-Assembly of Microtubule Tactoids
The following protocol is adapted from established visual experiments [2] [7].
1. Tubulin Preparation:
- Resuspend lyophilized unlabeled tubulin (1 mg) in 200 µL of cold PEM-80 buffer (80 mM PIPES, 1 mM EGTA, 4 mM MgCl₂, pH 6.9).
- Resuspend lyophilized Rhodamine-labeled tubulin (20 µg) in 4 µL of cold PEM-80.
- Combine 100 µL of the unlabeled tubulin solution with the 4 µL of Rhodamine-labeled tubulin. Mix gently by pipetting. Aliquot, flash-freeze in liquid nitrogen, and store at -80°C.
2. Flow Chamber Assembly:
- Clean a glass slide thoroughly with water and ethanol.
- Adhere two thin strips of double-sided tape to the slide, 5-8 mm apart.
- Place a silanized coverslip on top of the tape to create a sealed flow chamber.
- Press gently to ensure a complete seal.
3. Sample Preparation and Tactoid Formation:
- Coat the flow chamber with a 5% solution of a non-ionic block copolymer surfactant (e.g., Pluronic F-127) in PEM-80 for 5-7 minutes to passivate the surface.
- Prepare the final assembly mix on ice, containing:
- PEM-80 buffer
- GMPCPP (to stabilize microtubules)
- Pluronic F-127
- Dithiothreitol (DTT, a reducing agent)
- Glucose
- PEG (crowding agent)
- The prepared tubulin mix
- MAP65 (including a fraction tagged with GFP for visualization)
- Add glucose oxidase and catalase to the mix immediately before use.
- Introduce the mixture into the flow chamber via capillary action.
- Seal the chamber ends with epoxy and incubate at 37°C for 30 minutes to allow for microtubule nucleation and tactoid self-assembly.
FRAP Assay Execution
1. Microscope Setup:
- Use a confocal or TIRF microscope with a 60x or higher magnification objective (NA ≥ 1.2) and an environmental chamber maintained at 37°C.
- For Rhodamine-tubulin, use a 561 nm laser for excitation and a suitable emission filter (e.g., 575 nm long-pass).
- For GFP-MAP65, use a 488 nm laser and a corresponding emission filter (e.g., 515 nm long-pass) [7].
2. Photobleaching and Recovery Imaging:
- Identify a well-formed tactoid and select a rectangular region spanning its width for photobleaching.
- Acquire a few pre-bleach images with low laser intensity to record the initial fluorescence.
- Photobleach the selected region using a high-intensity pulse of the appropriate laser.
- Immediately begin time-lapse imaging at low laser power to track fluorescence recovery without causing further significant photobleaching.
- Continue imaging for a sufficient duration (e.g., 5-10 minutes) to capture the full recovery kinetics for MAP65.
Diagram 1: The sequential workflow for performing a FRAP experiment on a microtubule tactoid.
Quantitative Data Interpretation
The FRAP recovery curves for microtubules and MAP65 are fundamentally different, revealing their distinct physical states within the tactoid.
Table 2: Summary of FRAP results and their interpretation for microtubules and MAP65 in tactoids.
| Component | Fluorescence Recovery | Quantitative Recovery Parameters | Molecular Interpretation |
|---|---|---|---|
| Microtubules | No significant recovery observed after photobleaching [7]. | Mobile Fraction: ~0% [7]. | The microtubule lattice is static and immobile. Short, stable (GMPCPP-bound) filaments are crosslinked into a solid-like network without measurable subunit turnover or lateral diffusion [2] [7]. |
| MAP65 | Gradual recovery of fluorescence after photobleaching [7]. | Recovery time constant: Several seconds (fitted with a rising exponential) [7].Mobile Fraction: Significant (>0%). | MAP65 molecules are dynamic. They undergo continuous binding and dissociation from the microtubule bundle, allowing them to exchange freely with the surrounding solution and diffuse within the tactoid [7]. |
Diagram 2: A kinetic model for FRAP interpretation. For microtubules (green), the bleached subunits cannot be replaced, leading to no recovery. For MAP65 (yellow/orange), bleached molecules dissociate and are replaced by fluorescent ones from the pool, enabling fluorescence recovery.
Discussion and Application Notes
The observed FRAP dichotomy is a hallmark of a solid-like scaffold maintained by a dynamic crosslinking protein.
Immobile Microtubules Define a Stable Scaffold: The lack of microtubule fluorescence recovery confirms that the tactoid is not a liquid crystalline phase in this context but a static, solid bundle. This is a direct consequence of using GMPCPP-stabilized microtubules, which suppress the inherent dynamic instability of microtubules. The structure's integrity and shape are maintained by the mechanical strength of the crosslinked bundle rather than by liquid-like coalescence [2] [7].
Mobile MAP65 Enables Structural Plasticity: The recovery of MAP65 fluorescence demonstrates that the crosslinks holding the bundle together are transient. This dynamic binding is crucial for the self-organization and reorganization of the structure. It allows the tactoid to adjust to internal stresses and incorporate new subunits without dissolving the overall architecture. This behavior mirrors that of PRC1, the mammalian homolog of MAP65, which forms crosslinks that can be remodeled by motor proteins like Kif4A [22].
Broader Implications for Spindle Biology and Drug Discovery: The microtubule tactoid system, combined with FRAP analysis, serves as a minimal model for the spindle midzone. The solid microtubule core provides mechanical stability, while dynamic MAPs allow for regulatory inputs. For drug development, this system can be used to screen for compounds that modulate cytoskeletal stability. For instance, agents that stabilize or destabilize microtubules would be expected to lock or fluidize the tactoid structure, respectively, while compounds interfering with MAP65/PRC1 function would directly impact crosslink dynamics, which can be precisely quantified via FRAP.
The FRAP-based distinction between an immobile microtubule lattice and a mobile MAP65 population in reconstituted tactoids provides a powerful paradigm for deconstructing the biophysical principles of complex cellular assemblies. The detailed protocol and interpretive framework outlined here equip researchers to apply this minimal system to probe the mechanisms of spindle assembly, the function of microtubule-associated proteins, and the action of pharmacological agents targeting the cytoskeleton.
The mitotic spindle is a fundamental self-organized structure responsible for accurate chromosome segregation during cell division. A key challenge in cell biology has been to reconstitute this complex machinery in vitro to understand the minimal components required for its assembly and function. Recent advances have demonstrated that microtubules can be reconstituted into spindle-like assemblies known as tactoids using a minimal set of components [1] [2]. These tactoids are liquid crystalline droplets that nucleate and grow from an isotropic state, bearing a striking resemblance to the shape of meiotic spindles [2]. This application note provides a detailed comparative analysis of reconstituted microtubule tactoids and native meiotic spindles, offering structured protocols and data to guide research and drug discovery in cell division biology.
Structural and Functional Comparison
The following table summarizes the key similarities and differences between self-assembled microtubule tactoids and native meiotic spindles, highlighting their structural, compositional, and functional characteristics.
| Characteristic | Reconstituted Microtubule Tactoids | Native Meiotic Spindles (e.g., in Zea mays) |
|---|---|---|
| Overall Shape | Bipolar, tapered "spindle" or football shape; can be homogeneous (unipolar) or bipolar [2]. | Bipolar, barrel-like shape; described as a "pole-indented tactoid" [51]. |
| Primary Components | Tubulin, antiparallel crosslinkers (e.g., MAP65), crowding agents (e.g., PEG) [1] [2]. | Microtubules, kinetochore complexes, motor proteins (e.g., dynein, kinesins), MAPs, membranes [52] [53]. |
| Microtubule Organization | Short, crosslinked microtubules organized as a nematic liquid crystal [2]. | Defined classes: Kinetochore-MTs (kMTs), Interpolar-MTs, Astral-MTs; k-fibers contain 8–18 kMTs per kinetochore [52]. |
| Liquid Crystal Properties | Exhibits solid-like properties; mesoscale mesogens [2]. | Behaves as an active nematic liquid crystal; nematic elasticity governs structure and chromosome repulsion [51]. |
| Force Generation | Not explicitly measured; shape suggests internal stresses. | Active forces from molecular motors (e.g., dynein); long-range chromosome repulsion via nematic field deformation [51] [53]. |
| Key Crosslinker | MAP65 (plant homolog of PRC1/Ase1) [2]. | Multiple crosslinkers and motors, including PRC1 [2]. |
Experimental Protocol for Reconstituting Microtubule Tactoids
The following section details a standardized protocol for creating and analyzing microtubule tactoids in vitro.
Key Research Reagent Solutions
The table below lists the essential materials and their functions required for the successful assembly of microtubule tactoids.
| Research Reagent | Function/Explanation |
|---|---|
| Tubulin | The fundamental building block of microtubules; requires a high density of short filaments. GMPCPP is used to nucleate and stabilize short microtubules against dynamic instability [2]. |
| MAP65 | An antiparallel microtubule crosslinker from plants (homolog of Ase1/PRC1). It is a dimer that self-assembles and binds microtubules, essential for organizing short microtubules into tactoids [2]. |
| PEG (Polyethylene Glycol) | A crowding agent that creates depletion interactions, increasing the effective local concentration of microtubules and promoting their condensation into bundles and tactoids [2]. |
| Silanized Coverslips | Coverslips treated with dimethyldichlorosilane (DDS) to create a hydrophobic surface. This allows a block copolymer to bind and form a polymer brush, providing a passivated surface for assembly chambers [2]. |
Detailed Workflow
The protocol involves surface preparation, sample preparation, assembly, and characterization. The workflow is designed to ensure reproducible formation of tactoids for quantitative analysis.
Figure 1: Experimental workflow for the self-assembly and analysis of microtubule tactoids.
2.2.1 Coverslip Silanization
- Purpose: To create a hydrophobic surface on glass coverslips that enables the subsequent formation of a passivating polymer brush, preventing non-specific protein adhesion.
- Procedure:
- Clean: Rinse coverslips with ddH₂O, 70% ethanol, and ddH₂O again, drying with lint-free wipes between steps.
- UV-Ozone Clean: Place coverslips in a holder and irradiate in a UV-Ozone machine for 20 minutes to remove organic contaminants and background fluorescence.
- Solvent Rinse: Immerse coverslips in a rack sequentially in 100% acetone (1 hour), 100% ethanol (10 minutes), and ddH₂O (three times for 5 minutes each), rinsing the container thoroughly between steps.
- KOH Etch: Immerse coverslips in 0.1 M KOH for 15 minutes, then rinse the container and perform three more 5-minute ddH₂O rinses.
- Dry: Air-dry the rack and coverslips overnight in a fume hood.
- Silanize: In a fume hood, immerse the dried rack in 2% dimethyldichlorosilane (DDS) in a dedicated container for 5 minutes. Caution: DDS is highly toxic.
- Final Rinse: Rinse the silanized coverslips twice in 100% ethanol for 5 minutes each [2].
2.2.2 Tactoid Assembly Reaction
- Purpose: To combine the essential components in a controlled environment to promote the self-organization of microtubules into tactoids.
- Procedure:
- Prepare Tubulin: Use GMPCPP-stabilized tubulin to create a population of short, stable microtubules that resist dynamic instability and end-to-end annealing.
- Mix Components: Combine the following in an assembly buffer:
- Short, stabilized microtubules
- MAP65 crosslinking protein
- PEG (as a crowding agent)
- Chamber Assembly: Introduce the reaction mixture into a flow chamber constructed from the silanized coverslips and a passivated slide.
- Incubate: Allow the chamber to incubate at room temperature for the tactoids to form [2].
2.2.3 Characterization and Analysis
- Purpose: To quantify the physical and material properties of the assembled tactoids.
- Fluorescence Microscopy:
- Use fluorescence microscopy to image the tactoids.
- Quantify their shape parameters, such as length, width, and taper angle [2].
- Fluorescence Recovery After Photobleaching (FRAP):
Hierarchical Organization: From Molecules to Spindles
The diagram below illustrates the conceptual pathway from individual molecules to complex cellular structures, positioning reconstituted tactoids as an intermediate model system.
Figure 2: The hierarchical pathway of spindle assembly, from molecules to functional cellular structures.
Discussion and Application Notes
The comparative analysis reveals that reconstituted tactoids successfully capture essential physical aspects of meiotic spindles, particularly their bipolar, tactoid-shaped morphology governed by liquid crystal physics [2] [51]. The self-assembly protocol demonstrates that a minimal system of short microtubules, an antiparallel crosslinker (MAP65), and molecular crowding is sufficient to generate this defining shape.
However, key differences exist. Reconstituted tactoids lack the functional complexity of meiotic spindles, including the ability to segregate chromosomes. Native spindles are active nematic systems where energy from motor proteins like dynein generates internal stresses that further shape the spindle and organize its interior [53]. For example, inhibiting dynein in Xenopus egg extracts causes spindle poles to unfocus, demonstrating that active forces are required for the native structure [53]. Furthermore, the material properties differ; while tactoids can be solid-like, native spindles exhibit liquid-like properties that allow for chromosome re-orientation and long-range repulsive forces mediated by nematic elasticity [51].
For researchers, this protocol provides a robust platform for:
- Drug Discovery Screening: Tactoids can serve as a primary screen for compounds targeting microtubule integrity and bundling before testing in complex cellular assays.
- Mechanistic Studies: The system allows for direct testing of how specific MAPs or motors influence the emergent shape and mechanics of the assembly by adding them to the minimal reaction.
- Material Science of Active Nematics: Introducing molecular motors into tactoids could transition them from passive to active nematic systems, bridging the gap to native spindle behavior.
The protocol for self-assembling microtubule tactoids offers a powerful, reductionist model for studying the physical principles of spindle formation. While it does not fully replicate the biological complexity of the meiotic spindle, it provides unparalleled control for dissecting the role of core components like MAP65 and for probing the liquid crystal physics that underpin one of cell biology's most essential structures. This application note establishes a foundation for using this system in advanced research and development, from basic science to applied pharmacology.
Advantages and Limitations of the MAP65-based Minimal System
The MAP65-based minimal system represents a significant advancement in vitro reconstitution approaches for studying the self-organization of complex biological structures. By utilizing a minimal set of components—primarily microtubules, the plant-derived antiparallel crosslinker MAP65, and a crowding agent—this system spontaneously forms spindle-like assemblies known as microtubule tactoids [1] [20]. These structures are analogous to liquid crystal tactoids and provide a simplified model for investigating the physical principles underlying the formation of the mitotic spindle [20] [6]. This application note details the system's capabilities, constraints, and detailed methodologies, providing researchers with a framework for exploring cytoskeletal self-organization.
Cell division relies on the mitotic spindle, a transient microtubule-based structure that segregates chromosomes. The spindle's formation is a quintessential example of biological self-organization, where cellular components assemble without central direction [20]. A key protein family facilitating this process in plants is Microtubule-Associated Protein 65 (MAP65), which crosslinks microtubules in an antiparallel fashion [54]. MAP65 is a functional homolog of Ase1 in yeast and PRC1 in mammals [1] [20].
The minimal system reconstitutes this process in vitro using a limited number of purified components: short, stabilized microtubules, MAP65, and the crowding agent polyethylene glycol (PEG) [20] [7]. This system yields microtubule tactoids, which are bipolar, spindle-shaped assemblies that serve as a valuable model for dissecting the roles of molecular constituents and physical forces in shaping the metaphase spindle [20] [6].
Advantages of the MAP65-Based System
The MAP65-based system offers several distinct benefits for fundamental biophysical and cell biological research.
Simplified and Controlled Investigation
A primary advantage is the ability to isolate specific interactions in a well-defined environment, free from the complex regulatory network of a living cell [20] [7]. This simplification allows researchers to attribute observations directly to the known components—microtubules, MAP65, and crowders—enabling rigorous testing of hypotheses about spindle assembly.
Reconstitution of Spindle-Like Architecture
This minimal system successfully reconstitutes key structural features of the meiotic spindle. The resulting tactoids are long, thin, and tapered, mirroring the characteristic bipolar shape of the mitotic spindle [20] [6]. This demonstrates that the physical properties of microtubules, combined with a specific crosslinking protein, are sufficient to generate this complex morphology.
Model for Liquid Crystal Behavior
Microtubules in tactoids exhibit mesogen properties similar to molecules in liquid crystals [20] [6]. Their high aspect ratio and stiffness make them scaled-up versions of liquid crystal molecules, allowing the system to serve as a mesoscale model for studying the nucleation and growth of nematic phases from isotropic solutions. This provides a unique bridge between soft matter physics and cell biology.
Protein Mobility within Solid-Like Assemblies
A notable feature revealed by Fluorescence Recovery After Photobleaching (FRAP) is the differential mobility of components within the assembled tactoids. While the microtubule network itself is solid-like and shows no fluorescence recovery after bleaching, the MAP65 protein is mobile, gradually re-entering the bleached zone [7]. This indicates that the cross-links are dynamic, which could be crucial for the structural plasticity of the spindle.
Limitations and Technical Challenges
Despite its strengths, the system has several limitations that must be considered when interpreting results.
Absence of Motor Proteins and Enzymatic Regulation
The system notably lacks motor proteins (e.g., kinesins, dyneins) and enzymes that regulate microtubule dynamics [20]. These components are essential in living cells for generating forces to move chromosomes, focus spindle poles, and regulate microtubule length. Their absence limits the system's ability to fully model the dynamic processes of anaphase or the transport of cellular cargo [20].
Solid-Like versus Liquid-Like Properties
In vivo, meiotic spindles display liquid-like properties, allowing them to coalesce [20]. In contrast, microtubule tactoids formed with MAP65 are reported to be solid-like [20] [7]. This difference in material property suggests that additional cellular factors are necessary to confer the fluidity observed in living systems.
Critical Dependence on Microtubule Length
The successful formation of tapered tactoids requires the use of short microtubules [20]. Protocols must use guanosine-5'-[(α,β)-methyleno]triphosphate (GMPCPP) to nucleate and stabilize short filaments, preventing them from undergoing dynamic instability or end-to-end annealing [20]. The use of long microtubules often results in bundles without the desired spindle-like taper [20].
Technical Complexity and Sensitivity
The protocol involves several sensitive and time-critical steps. For instance, the tubulin used in the tactoid experiment can degrade quickly on ice, and the entire preparation and loading process must be completed within 10-12 minutes to ensure successful nucleation [7]. Furthermore, coverslip silanization requires handling highly toxic chemicals and meticulous cleaning procedures [20].
Table 1: Key Advantages and Limitations of the MAP65-Based Minimal System
| Aspect | Advantages | Limitations |
|---|---|---|
| System Complexity | Simplified, controlled environment for probing specific interactions [20] [7]. | Lacks motor proteins, regulatory enzymes, and other cellular factors [20]. |
| Structural Output | Reconstitutes bipolar, spindle-shaped tactoids [20] [6]. | Assemblies are solid-like, unlike more liquid-like meiotic spindles in vivo [20] [7]. |
| Physical Insights | Serves as a mesoscale model for liquid crystal behavior [20] [6]. | Requires short microtubules; long filaments form bundles, not tactoids [20]. |
| Component Dynamics | Reveals mobile crosslinkers (MAP65) within a stable microtubule network [7]. | Technically challenging protocol with sensitive, time-critical steps [20] [7]. |
Experimental Protocol for Microtubule Tactoid Assembly
The following section provides a detailed methodology for forming and analyzing microtubule tactoids, as derived from established visual protocols [20] [7].
Preparation of Silanized Coverslips
Coverslips must be treated to create a hydrophobic surface for the subsequent polymer brush coating.
- Clean Coverslips: Rinse coverslips sequentially with ddH₂O, 70% ethanol, and ddH₂O again, drying with lint-free wipes between steps [20].
- UV-Ozone Treatment: Place coverslips in a holder and irradiate in a UV-Ozone machine for 20 minutes to remove background fluorescence [20].
- Solvent Rinses: Immerse coverslips in a rack through a series of rinses: 1 hour in 100% acetone, 10 minutes in 100% ethanol, and three 5-minute rinses in ddH₂O [20].
- Alkaline Clean: Immerse in 0.1 M KOH for 15 minutes, followed by three 5-minute ddH₂O rinses [20].
- Air Dry: Let the rack and coverslips air-dry overnight in a fume hood [20].
- Silanization: Immerse dried coverslips in 2% dimethyldichlorosilane (DDS) in a dedicated container for 5 minutes. Caution: DDS is highly toxic; use a fume hood and gloves [20].
- Final Rinsing and Drying: Perform two 5-minute rinses in 100% ethanol, followed by three 5-minute rinses in ddH₂O [20].
- Air Dry overnight, then store the treated coverslips in a clean box for up to two months [20].
Tubulin Preparation and Labeling
This protocol prepares a tubulin mixture suitable for fluorescence visualization.
- Resuspend Unlabeled Tubulin: Obtain a 1 mg aliquot of unlabeled, lyophilized tubulin from the -80°C freezer. Keep on ice and add 200 µL of cold PEM-80 buffer to achieve a 5 mg/mL concentration. Hold on ice for 10 minutes to dissolve [20] [7].
- Resuspend Labeled Tubulin: Obtain a 20 µg aliquot of rhodamine-labeled, lyophilized tubulin. Add 4 µL of cold PEM-80 buffer and keep on ice for 10 minutes to dissolve [7].
- Mix Tubulin: Combine 100 µL of the unlabeled tubulin solution with the entire 4 µL of rhodamine-labeled tubulin solution. Mix by pipetting slowly 6-7 times [7].
- Aliquot and Store: Distribute the mix into seven tubes (15 µL each), flash-freeze in liquid nitrogen, and store at -80°C for future use [7].
Flow Chamber Assembly
- Clean Slide: Clean a glass slide with ddH₂O and ethanol, then dry it [7].
- Create Flow Path: Split a 40-50 mm piece of double-sided tape into two thin strips. Place them on the slide 5-8 mm apart [7].
- Attach Coverslip: Place a silanized coverslip on the tape strips to form a tunnel. Press gently with a pen back to seal, turning the tape from translucent to clear [7].
- Trim Tape: Use a razor blade to trim excess tape, leaving about 1 mm at the chamber entrance [7].
Tactoid Formation Experiment
This is a time-sensitive procedure; all reagents should be thawed and kept on ice.
- Coat Chamber: Introduce 20 µL of a 5% non-ionic block copolymer surfactant (e.g., Pluronic F-127) in PEM-80 into the flow chamber. Incubate in a humid chamber for 5-7 minutes [7].
- Prepare Reaction Mix: In a sterile tube on ice, combine the following components by pipetting 5-6 times [7]:
- PEM-80 buffer
- GMPCPP (nucleotide)
- Pluronic F-127
- Dithiothreitol (DTT)
- Glucose
- Polyethylene Glycol (PEG)
- Thawed tubulin mix
- MAP65 (with a fraction being GFP-MAP65 for visualization)
- Add Oxygen Scavengers: Add 1 µL of a pre-mixed glucose oxidase and catalase solution to the tubulin-MAP65 mix. Pipette 7-8 times to mix [7].
- Load Chamber: Remove the surfactant from the flow chamber by capillary action with a lint-free wipe. Immediately add the tubulin-MAP65 reaction mix to the chamber [7].
- Seal and Incubate: Seal both ends of the chamber with five-minute epoxy. Incubate at 37°C for 30 minutes for tactoids to nucleate and grow [7].
Imaging and Analysis
- Microscopy Setup: Use a microscope with a 60x or higher magnification objective (numerical aperture ≥1.2) and an environmental chamber set to 37°C [7].
- Fluorescence Imaging: For rhodamine-tubulin, use a 561 nm laser. For GFP-MAP65, use a 488 nm laser. Acquire at least 10 images from different areas to capture over 100 tactoids [7].
- FRAP Analysis: To probe dynamics, perform Fluorescence Recovery After Photobleaching. Bleach a region within a tactoid and monitor recovery over time. MAP65 will show recovery, while microtubules will not [7].
- Morphometric Analysis: Measure the length and width of tactoids from acquired images to quantify assembly shapes [7].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for MAP65-based Tactoid Assembly
| Reagent | Function/Description | Key Details |
|---|---|---|
| Tubulin | Core structural protein; polymerizes to form microtubules. | Use short microtubules stabilized with GMPCPP [20]. A mix of unlabeled and rhodamine-labeled allows visualization [7]. |
| MAP65 | Antiparallel microtubule crosslinker. | Crosslinks microtubules to form the tactoid's bipolar structure [1] [20]. GFP-tagged version allows live imaging [7]. |
| Polyethylene Glycol (PEG) | Crowding agent. | Mimics the crowded cellular environment by creating depletion forces that promote microtubule bundling and condensation [20] [6]. |
| GMPCPP | Non-hydrolyzable GTP analog. | Used to nucleate and stabilize microtubules, preventing dynamic instability and ensuring they remain short for tactoid formation [20]. |
| Pluronic F-127 | Non-ionic surfactant. | Coats the flow chamber to prevent non-specific adhesion of proteins to the glass surface [7]. |
| Glucose Oxidase/Catalase | Oxygen-scavenging system. | Reduces photodamage during fluorescence imaging by removing oxygen from the solution [7]. |
Experimental Workflow and System Logic
The following diagrams illustrate the procedural flow of the experiment and the core logical relationships within the minimal system.
Tactoid Assembly Workflow
MAP65 System Logic
The MAP65-based minimal system is a powerful reductionist tool that has provided profound insights into the self-organizing principles of the cytoskeleton. Its ability to reconstitute a spindle-like structure with a minimal set of components underscores the importance of basic physical forces and specific protein interactions in cellular organization. While the system has inherent limitations, such as the absence of motor proteins and its solid-like material properties, these very constraints help define the essential requirements for more complex cellular processes. Future work integrating additional components like motor proteins and regulatory enzymes will build upon this foundational system to create increasingly sophisticated and dynamic models of intracellular assemblies.
Conclusion
The self-assembly of microtubule tactoids provides a powerful, minimalistic, and highly reproducible system to dissect the physical and biochemical principles underlying the organization of complex biological structures like the mitotic spindle. The successful implementation of this protocol hinges on understanding the foundational liquid crystal concepts, meticulously following the preparation steps, and proactively applying troubleshooting knowledge, particularly regarding the critical impact of ionic strength. The ability to characterize these assemblies through fluorescence microscopy and FRAP offers profound insights into the dynamics and material state of the cytoskeleton. Future directions for this research include integrating other cytoskeletal components and motor proteins to create more complex and dynamic synthetic cellular systems. For biomedical research, this protocol establishes a foundational platform for high-throughput screening of drugs that target microtubule networks, with potential implications for understanding cell division errors and developing next-generation chemotherapeutics.
References
- 1. Self-Assembly of Microtubule Tactoids - PubMed [https://pubmed.ncbi.nlm.nih.gov/35815974/]
- 2. Self-Assembly of Microtubule Tactoids [https://www.jove.com/t/63952/self-assembly-of-microtubule-tactoids]
- 3. Investigating mitotic assembly and function spindle using... in vitro [https://www.nature.com/articles/nprot.2006.396?error=cookies_not_supported&code=042aceb9-39cf-4762-9858-c2bfe17f7f0d]
- 4. assembly by two different pathways Mitotic . | Scilit spindle in vitro [https://www.scilit.com/publications/ac68de5ea7dc2b929c255ef71a6adf01]
- 5. (PDF) Physiological and ultrastructural analysis of elongating mitotic ... [https://www.academia.edu/56794902/Physiological_and_ultrastructural_analysis_of_elongating_mitotic_spindles_reactivated_in_vitro]
- 6. (PDF) Self-Assembly of Microtubule Tactoids | Prashali Chauhan - Academia.edu [https://www.academia.edu/118355287/Self_Assembly_of_Microtubule_Tactoids]
- 7. Video: Self-Assembly of Microtubule Tactoids [https://www.jove.com/v/63952/self-assembly-of-microtubule-tactoids]
- 8. : ordered Liquid , defective coalescence... crystalline tactoids structure [https://pmc.ncbi.nlm.nih.gov/articles/PMC5746557/]
- 9. - Wikipedia Tactoid [https://en.wikipedia.org/wiki/Tactoid]
- 10. in microscopic ordered-disordered... Liquid crystalline tactoids [https://open.library.ubc.ca/soa/cIRcle/collections/ubctheses/24/items/1.0372765]
- 11. Shape and relaxation of colloidal structural tactoids [https://www.nature.com/articles/s41467-022-30123-y?error=cookies_not_supported&code=4f6a2fbe-7d90-4efe-9f7d-08fcd3b043b5]
- 12. - Self of Assembly Microtubule Tactoids [https://www.jove.com/pl/t/63952/self-assembly-of-microtubule-tactoids]
- 13. Flow-induced order–order transitions in amyloid fibril liquid ... crystalline [https://www.nature.com/articles/s41467-020-19213-x?error=cookies_not_supported&code=67c28fe0-660b-461f-a4e1-7367f52b664a]
- 14. Rapid Microtubule Self-assembly Kinetics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3171214/]
- 15. Mechanisms of the Ase / 1 / PRC family in central spindle... 1 MAP 65 [https://pubmed.ncbi.nlm.nih.gov/31343816/]
- 16. Interplay of self-organization of microtubule asters and crosslinking protein condensates (Journal Article) | NSF PAGES [https://par.nsf.gov/biblio/10517105-interplay-self-organization-microtubule-asters-crosslinking-protein-condensates]
- 17. Macromolecular Crowding Pushes Catalyzed Microtubule Growth to... [https://link.springer.com/article/10.1007/s12195-013-0292-9]
- 18. Effects of microtubule length and crowding on active microtubule... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9958361/]
- 19. Live visualizations of single isolated tubulin protein self-assembly via tunneling current: effect of electromagnetic pumping during spontaneous growth of microtubule | Scientific Reports [https://www.nature.com/articles/srep07303]
- 20. - Self of Assembly Microtubule Tactoids [https://app.jove.com/t/63952/self-assembly-of-microtubule-tactoids]
- 21. INSIGHTS INTO ANTI-PARALLEL MICROTUBULE CROSSLINKING BY PRC1, A CONSERVED NON-MOTOR MICROTUBULE BINDING PROTEIN - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2966277/]
- 22. Geometry of antiparallel bundles regulates relative... microtubule [https://elifesciences.org/articles/32595]
- 23. Kinesin-14 motors drive a right-handed helical motion of antiparallel ... [https://www.nature.com/articles/s41467-020-16328-z?error=cookies_not_supported&code=acff4aa2-405d-4c00-a98a-cf2f3074b11b]
- 24. Simultaneous Visualization of the Dynamics of Crosslinked and Single... [https://www.jove.com/t/63377/simultaneous-visualization-dynamics-crosslinked-single-microtubules]
- 25. Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau | Nature Communications [https://www.nature.com/articles/s41467-017-02230-8]
- 26. Cross-linkers at growing microtubule ends generate forces that drive actin transport - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8931237/]
- 27. – Kinesin Tubulin Preparation [https://sites.duke.edu/kinesin/tubulin_prep/]
- 28. Large Scale Tubulin Preparation [https://diyhpl.us/~bryan/irc/protocol-online/protocol-cache/tubprep.html]
- 29. Interface-acting nucleotide controls polymerization dynamics at... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10187237/]
- 30. Double symmetry breaking in filamentous colloidal tactoids [https://pubs.rsc.org/en/content/articlehtml/2025/sm/d5sm00236b]
- 31. - Self of Assembly - JoVE Microtubule Tactoids [https://www.jove.com/es/t/63952/self-assembly-of-microtubule-tactoids/]
- 32. alters crosslinker-driven Ionic -organization of... strength self [https://pubmed.ncbi.nlm.nih.gov/38385864/]
- 33. Supramolecular polymers form tactoids through liquid–liquid phase separation | Nature [https://www.nature.com/articles/s41586-024-07034-7]
- 34. - Wikipedia Microtubule [https://en.wikipedia.org/wiki/Microtubule]
- 35. Insights into the mechanism of microtubule by Taxol... stabilization [https://pmc.ncbi.nlm.nih.gov/articles/PMC1502429/]
- 36. General Tubulin Buffer - Cytoskeleton, Inc. [https://www.cytoskeleton.com/bst01]
- 37. Influence of M-phase chromatin on the anisotropy of microtubule asters - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2120784/]
- 38. The transition state and regulation of γ-TuRC-mediated microtubule ... [https://elifesciences.org/articles/54253]
- 39. sciencedirect.com/topics/medicine-and-dentistry/ microtubule ... [https://www.sciencedirect.com/topics/medicine-and-dentistry/microtubule-nucleation]
- 40. XMAP215 is a microtubule that functions... nucleation factor [https://pubmed.ncbi.nlm.nih.gov/29695792/]
- 41. XMAP215 is a microtubule ... | Nature Cell Biology nucleation factor [https://www.nature.com/articles/s41556-018-0091-6?error=cookies_not_supported&code=5907ce88-64a7-4b7b-a8f1-9e91620bd077]
- 42. and Surface Crowder on Self-organization of Effects Microtubules [https://arxiv.org/abs/2009.04669]
- 43. Nonlinear Methods for Shape Optimization Problems in Liquid Crystal Tactoids [https://arxiv.org/html/2310.04022]
- 44. Measuring microtubule dynamics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6281472/]
- 45. Autoensamblaje de tactoides de microtúbulos [https://www.jove.com/es/t/63952]
- 46. of Quantitative - analysis kinetics and tip... microtubule self assembly [https://pubmed.ncbi.nlm.nih.gov/24630100/]
- 47. for observing Protocol and microtubules ends in both... microtubule [https://pmc.ncbi.nlm.nih.gov/articles/PMC10448205/]
- 48. - Wikipedia Fluorescence recovery after photobleaching [https://en.wikipedia.org/wiki/Fluorescence_recovery_after_photobleaching]
- 49. of Junctional Protein Dynamics Using Measurement ... Fluorescence [https://bio-protocol.org/en/bpdetail?id=937&type=0]
- 50. (Fluorescence Recovery After Photobleaching) - FluoroFinder FRAP [https://fluorofinder.com/frap-fluorescence/]
- 51. Long-range repulsion between chromosomes in mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11423871/]
- 52. Zea mays Meiotic Spindle Ultrastructure Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503593/]
- 53. Active forces shape the metaphase spindle through a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7368317/]
- 54. Comprehensive analyses of microtubule-associated protein MAP ... 65 [https://pubmed.ncbi.nlm.nih.gov/37219731/]