A Complete Guide to LIMAP Light-Induced Molecular Adsorption Protein Micropatterning: Protocol, Applications & Troubleshooting
This comprehensive guide provides researchers, scientists, and drug development professionals with an in-depth protocol for Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning.
A Complete Guide to LIMAP Light-Induced Molecular Adsorption Protein Micropatterning: Protocol, Applications & Troubleshooting
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with an in-depth protocol for Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning. It details the foundational principles behind this powerful, non-contact technique for creating precise, biologically active protein patterns on surfaces. We cover a step-by-step, optimized methodological workflow, including substrate preparation, photomask design, light activation parameters, and protein conjugation. Critical troubleshooting and optimization strategies address common pitfalls to ensure reproducibility. Finally, we validate the technique by comparing LIMAP to alternative patterning methods (e.g., microcontact printing, inkjet printing) and demonstrating its applications in single-cell studies, high-content screening, and engineered tissue models. This guide serves as an essential resource for implementing robust and versatile surface patterning in biomedical research.
What is LIMAP Micropatterning? Understanding the Core Principles and Advantages
Article Context
This article presents foundational protocols and application notes within a broader thesis investigating Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning. This research aims to establish a robust, high-resolution platform for spatially controlling biomolecular interactions on surfaces, with direct applications in fundamental cell biology, biosensor development, and targeted drug discovery.
LIMAP utilizes photoresponsive molecules (e.g., photochromic azobenzenes, coumarins) anchored to a substrate. Upon illumination with specific wavelengths, these molecules undergo reversible conformational or polarity changes, inducing the adsorption or desorption of target proteins from solution with precise spatiotemporal control.
Table 1: Quantitative Performance Metrics of Recent LIMAP Systems
| System Component | Key Metric | Typical Reported Value (Range) | Reference Year | Notes |
|---|---|---|---|---|
| Spatial Resolution | Minimum Feature Size | 1 - 5 µm | 2023 | Dependent on optics, photomask, and linker chemistry. |
| Temporal Control | Adsorption Onset Time | < 5 seconds | 2024 | Time from illumination to detectable protein binding. |
| Reversibility | Cycle Stability (Adsorption/Desorption) | > 20 cycles | 2023 | Maintains >80% of initial binding capacity. |
| Protein Binding Density | Surface Coverage | 200 - 500 ng/cm² | 2024 | For fibronectin; varies by protein and surface chemistry. |
| Wavelength Dependency | Activation/Deactivation λ | 365 nm / 450 nm | 2023-2024 | Common for azobenzene-based systems. |
| Background Adsorption | Non-Irradiated Area Signal | < 5% of patterned area | 2024 | Critical for pattern fidelity. |
Detailed Protocols
Protocol 1: Substrate Preparation & Photolinker Functionalization
Objective: To create a gold or glass substrate uniformly coated with a photoresponsive silane linker.
- Substrate Cleaning: Sonicate glass coverslips in acetone, ethanol, and deionized water (10 min each). Dry under N₂ stream. For gold substrates, use piranha solution (Caution: Highly corrosive) followed by thorough rinsing.
- Oxygen Plasma Treatment: Treat clean substrates in a plasma cleaner for 5 minutes to generate surface hydroxyl groups.
- Photolinker Deposition: Incubate substrates in a 1 mM solution of 4-((3-(triethoxysilyl)propyl)diazenyl)benzoic acid (or equivalent azobenzene-trialkoxysilane) in anhydrous toluene for 12 hours under inert atmosphere.
- Washing & Curing: Rinse sequentially with toluene, ethanol, and water. Cure at 120°C for 1 hour to complete silanization.
- Quality Control: Verify monolayer formation by measuring water contact angle (expected ~70°) and by X-ray photoelectron spectroscopy (XPS) for nitrogen signature.
Protocol 2: Light-Induced Patterning of Extracellular Matrix Proteins
Objective: To create a defined micropattern of fibronectin on a prepared LIMAP substrate.
- Setup: Mount functionalized substrate in a flow chamber. Connect to a syringe pump.
- Protein Solution Preparation: Prepare a 50 µg/mL solution of fibronectin in PBS (pH 7.4).
- Illumination & Adsorption:
- Flush chamber with PBS.
- Using a digital micromirror device (DMD) or a photomask aligned to the substrate, illuminate the desired pattern with 365 nm UV light (intensity: 5-10 mW/cm²) for 60 seconds.
- Immediately introduce the fibronectin solution and incubate in the dark for 15 minutes.
- Deactivation & Washing:
- Illuminate the entire substrate with 450 nm visible light for 120 seconds to deactivate unpatterned areas.
- Flush chamber extensively with PBS containing 0.1% Tween-20 to remove non-specifically adsorbed protein.
- Validation: Stain with fluorescently labeled anti-fibronectin antibody and image via fluorescence microscopy. Quantify pattern fidelity and protein density.
Protocol 3: Cell Seeding on LIMAP Patterns
Objective: To seed cells that adhere exclusively to the protein-micropatterned regions.
- Blocking: Incubate the patterned substrate with 1% bovine serum albumin (BSA) in PBS for 1 hour to block non-specific cell adhesion.
- Cell Preparation: Trypsinize and resuspend NIH/3T3 fibroblasts (or relevant cell line) in serum-free medium at 1 x 10⁵ cells/mL.
- Seeding: Seed cell suspension onto the substrate and allow to adhere for 30-60 minutes in a humidified incubator (37°C, 5% CO₂).
- Washing: Gently rinse with PBS to remove non-adherent cells.
- Culture: Add complete growth medium and continue culture as required. Monitor confinement to patterned regions over 24-48 hours.
Visualizations
Title: LIMAP Experimental Workflow for Cell Patterning
Title: Molecular Mechanism of LIMAP Patterning and Erasure
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for LIMAP
| Item | Function in LIMAP Protocol | Key Specification/Note |
|---|---|---|
| Photoresponsive Silane (e.g., Azobenzene-trialkoxysilane) | Forms the photo-switchable monolayer on the substrate. | Purity >95%; requires anhydrous conditions for coupling. |
| Anhydrous Toluene | Solvent for silanization reaction. | Must be dry (<50 ppm H₂O) to prevent silane polymerization. |
| Target Protein (e.g., Fibronectin, Collagen I) | The biological molecule to be patterned. | Fluorescent labeling or tag may be required for visualization. |
| PBS with Tween-20 (0.1%) | Wash buffer to remove loosely adsorbed protein. | Reduces non-specific background adsorption. |
| Blocking Agent (e.g., BSA, Casein) | Passivates non-patterned areas to prevent non-specific cell adhesion. | Use protein-free buffers for subsequent specific binding assays. |
| Digital Micromirror Device (DMD) | Provides dynamic, high-resolution spatial control of activating light. | Enables complex, multi-pattern designs without physical masks. |
| LED Light Sources (365 nm & 450 nm) | Provides precise wavelength control for isomerization. | Intensity stability and uniformity are critical for reproducible adsorption. |
| Fluorescent Conjugates/Antibodies | For visualization and quantification of patterned protein. | Use secondary antibodies if primary is unlabeled. |
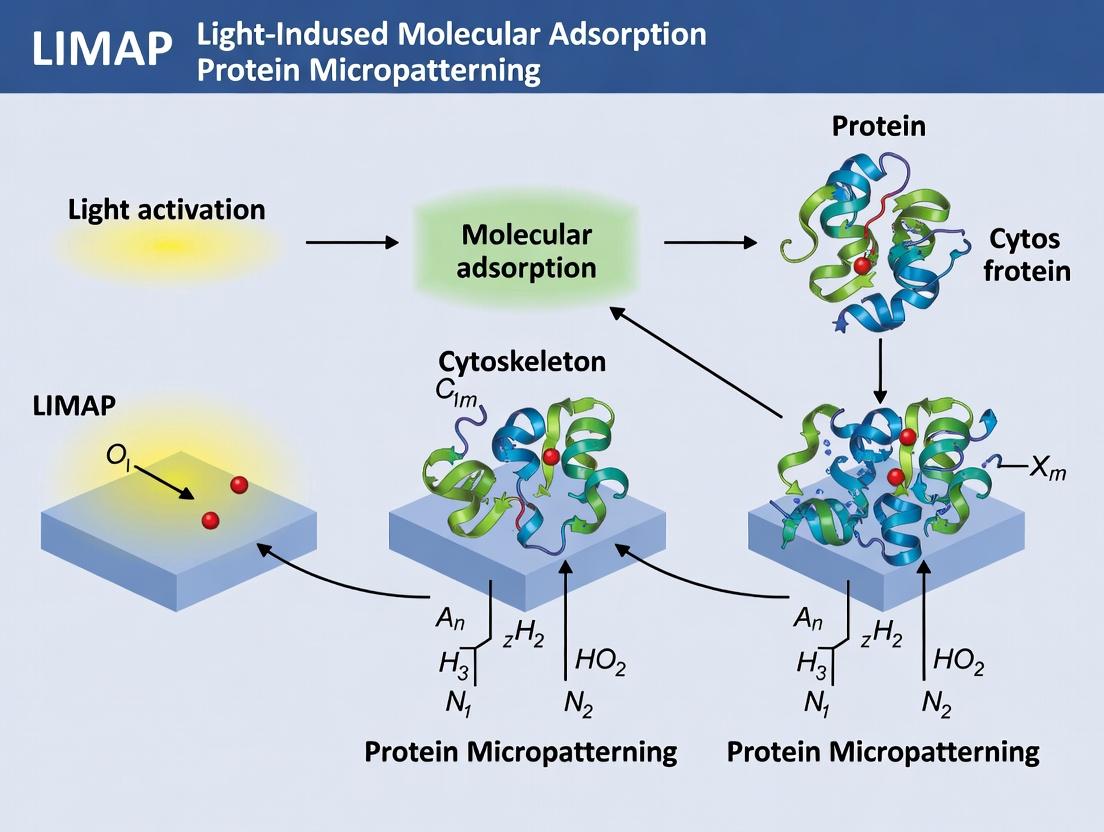
Within the broader thesis on LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning), this document details the core photochemical mechanism by which controlled light exposure renders inert surfaces protein-adhesive. LIMAP is a non-lithographic, spatiotemporally precise technique for creating complex protein patterns on surfaces like polystyrene or functionalized glass. The core mechanism involves the photolytic generation of reactive species from a photoactivatable coating, leading to the covalent or high-affinity adsorption of target proteins exclusively in illuminated zones.
Core Photochemical Mechanism
The standard LIMAP protocol employs a photoactivatable heterobifunctional crosslinker, Sulfo-SANPAH (Sulfosuccinimidyl 6-[4'-azido-2'-nitrophenylamino]hexanoate), coated onto the substrate.
Mechanistic Steps:
- Photon Absorption: Controlled UV-A light (~320-365 nm) is projected onto the Sulfo-SANPAH-coated surface in a defined pattern.
- Nitrene Generation: The aryl azide moiety of Sulfo-SANPAH absorbs photons, causing the loss of nitrogen (N₂) and generating a highly reactive aryl nitrene radical.
- Surface Activation: The nitrene inserts into C-H or N-H bonds of the underlying polymer substrate (e.g., polystyrene), covalently tethering the crosslinker to the surface.
- Protein Binding Site Presentation: Concurrently or subsequently, the NHS-ester end of the now surface-bound crosslinker hydrolyzes or reacts with amine groups (-NH₂) from the target protein in solution, forming a stable amide bond.
- Pattern Formation: Protein binding occurs only where light exposure created the activated nitrene, resulting in a high-fidelity protein micropattern.
Key Data & Experimental Parameters
Table 1: Quantitative Parameters for Effective LIMAP Surface Activation
| Parameter | Optimal Range / Value | Effect / Rationale |
|---|---|---|
| Wavelength | 320 - 365 nm (UV-A) | Peak absorption for aryl azide photolysis. Minimizes protein-damaging UV-B/C. |
| Light Dose | 0.5 - 2.0 J/cm² | Balance between complete azide conversion and substrate/protein damage. |
| Sulfo-SANPAH Concentration | 0.1 - 0.5 mg/mL in buffer | Ensures monolayer coverage without excessive multilayer aggregation. |
| Irradiation Buffer | Non-amine PBS or HEPES | Prevents quenching of nitrene by competitive amines. |
| Post-Irradiation Incubation | 4°C, 12-16 hours (Protein) | Allows efficient NHS-ester coupling to protein amines with minimal denaturation. |
| Pattern Resolution | ~1 µm (diffraction-limited) | Determined by optical system, wavelength, and photomask feature size. |
Table 2: Comparison of Photoactivatable Coating Chemistries
| Coating / Crosslinker | Reactive Species | Target on Substrate | Protein Coupling Chemistry | Key Advantage |
|---|---|---|---|---|
| Sulfo-SANPAH | Aryl Nitrene | C-H/N-H bonds (Polystyrene) | NHS-ester to -NH₂ | Well-established, reliable. |
| Benzophenone-derivatives | Triplet-state Ketone | C-H bonds | Varied (Acrylate, NHS) | More selective, less quenched by water. |
| Diazirine-derivatives | Carbene | Broad (C-H, O-H, N-H) | Varied | Smaller footprint, higher reactivity. |
Detailed Experimental Protocols
Protocol 4.1: Substrate Preparation & Coating
Objective: Apply a uniform layer of photoactivatable crosslinker onto a sterile cell culture dish.
- Use a 35 mm polystyrene cell culture dish.
- Prepare a fresh solution of 0.2 mg/mL Sulfo-SANPAH in sterile, non-amine PBS (pH ~7.4). Protect from light.
- Add 1.5 mL of the solution to cover the dish surface.
- Incubate for 30 minutes at room temperature in the dark.
- Aspirate the solution and rinse the dish gently three times with non-amine PBS to remove unbound crosslinker.
- Leave a thin film of PBS to keep the surface hydrated. Proceed immediately to patterning.
Protocol 4.2: Light Patterning & Protein Immobilization
Objective: Activate defined surface regions via UV exposure and subsequently adsorb the protein of interest.
- Setup: Place a high-resolution photomask (chrome on quartz) defining the desired pattern in direct contact with or projected onto the coated dish surface.
- Irradiation: Expose the dish to collimated UV-A light (e.g., 365 nm LED source) at an intensity of 10 mW/cm² for 100 seconds (Total Dose = 1.0 J/cm²).
- Rinse: Immediately after exposure, gently rinse the dish with patterning buffer (PBS).
- Protein Application: Incubate with a solution of the target protein (e.g., 10 µg/mL fibronectin in PBS) at 4°C for 16 hours. Protect from light.
- Quenching & Sterilization: Aspirate protein solution. Incubate with 1 mL of 1% BSA or 100 mM glycine in PBS for 1 hour to quench any remaining active esters. Rinse with PBS. For cell culture, sterilize under UV-C in a biosafety cabinet for 20 minutes.
- Validation: Pattern fidelity can be validated by incubating with fluorescently labeled protein (or antibody) and imaging via fluorescence microscopy.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for LIMAP Protocols
| Item | Function / Role in LIMAP | Example Product / Specification |
|---|---|---|
| Sulfo-SANPAH | Heterobifunctional photoreactive crosslinker. NHS-ester couples to protein amines; aryl azide photolyzes to bind surface. | Thermo Fisher Scientific, #22589 |
| Polystyrene Dishes | Inert substrate with abundant C-H bonds for nitrene insertion. | Tissue Culture Treated, 35mm, # glass bottom for imaging |
| Non-Amine PBS | Irradiation buffer. Absence of amines prevents quenching of the reactive nitrene species. | 10X Solution, pH 7.4 (e.g., Thermo Fisher, #28372) |
| UV-A Light Source | Provides controlled 365 nm light for photolysis. Must be collimated for sharp patterns. | High-power 365 nm LED array with condenser lens |
| High-Resolution Photomask | Defines the spatial pattern of light exposure. | Chrome-on-quartz mask, 1-20 µm features |
| Target Protein | Molecule to be patterned. Must contain primary amines (Lysine residues or N-terminus). | Fibronectin, Collagen, Laminin, Antibodies |
| Quenching Agent | Blocks unreacted NHS-ester groups after patterning to prevent non-specific binding. | 1% Bovine Serum Albumin (BSA) or 100 mM Glycine |
Visualizing the Mechanism & Workflow
Diagram 1: LIMAP Core Workflow
Diagram 2: Photochemical Activation Mechanism
This document provides application notes and protocols to support the broader thesis research on Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning. LIMAP represents a transformative approach to creating precise, high-fidelity protein patterns on surfaces, which is critical for advanced cell biology studies, tissue engineering, and drug development platforms. The core advantages of LIMAP—its non-contact nature, high resolution, and material versatility—directly address limitations inherent in traditional patterning techniques like microcontact printing (µCP) and inkjet printing, enabling novel experimental paradigms in biomaterial fabrication.
Core Advantages: Quantitative Comparison
Table 1: Comparative Analysis of Patterning Techniques
| Feature / Metric | Traditional µCP | Inkjet Printing | LIMAP (This Work) |
|---|---|---|---|
| Pattern Resolution | 1 - 50 µm | 20 - 100 µm | < 1 µm (demonstrated: 0.8 µm) |
| Contact with Substrate? | Yes (stamp contact) | No (droplet-based) | No (pure optical projection) |
| Typical Throughput | Medium (batch) | High (serial) | Medium-High (parallel projection) |
| Multiplexing Capability | Low (sequential stamping) | Medium (multi-nozzle) | High (dynamic mask, multi-wavelength) |
| Substrate Versatility | Low (flat, uniform) | Medium (low surface tension) | High (any photoadsorbent coating) |
| Feature Alignment Accuracy | ± 5 µm | ± 10-20 µm | ± 0.5 µm (stage-locked) |
| Protein Activity Retention | 60-80% (due to drying/transfer) | 50-70% (shear stress) | >90% (gentle in-situ adsorption) |
| Lateral Diffusion / Blurring | High (stamp bleeding) | Medium (droplet spreading) | Negligible (sharp optical boundary) |
Application Notes & Detailed Protocols
Protocol: High-Resolution Multi-Protein Line Pattern for Axon Guidance Studies
- Objective: Create alternating 2 µm lines of Laminin and Poly-L-Lysine (PLL) to study neuronal pathfinding.
- Advantage Leveraged: High Resolution & Non-Contact.
Materials & Reagents:
- Photosensitive Silane-coated coverslip (e.g., Azido-silane).
- Protein Solutions: Laminin (50 µg/mL in PBS), Biotinylated PLL (100 µg/mL).
- LIMAP System with Digital Micromirror Device (DMD) projector (365 nm LED).
- Phosphate Buffered Saline (PBS), pH 7.4.
- Blocking Solution: 1% Bovine Serum Albumin (BSA) in PBS.
Procedure:
- Substrate Activation: Mount photosensitive coverslip in LIMAP flow chamber. Prime with PBS.
- First Pattern Exposure: Flow Laminin solution into chamber. Project first line pattern (2 µm lines, 4 µm spacing) for 60 seconds at 10 mW/cm².
- Rinse: Flush chamber with 5 mL PBS to remove non-adsorbed Laminin.
- Blocking: Incubate with BSA blocking solution for 15 min to passivate non-patterned areas.
- Second Pattern Exposure: Flow Biotinylated PLL solution. Project the complementary line pattern (offset by 3 µm) for 45 seconds.
- Final Rinse: Flush with 10 mL PBS. The substrate is ready for cell seeding.
Protocol: 3D Hydrogel Patterning via LIMAP-Induced Crosslinking
- Objective: Generate graded patterns of cell-adhesive RGD peptide within a PEG-DA hydrogel.
- Advantage Leveraged: Versatility (non-planar, 3D materials).
Materials & Reagents:
- Precursor Solution: 10% (w/v) Polyethylene Glycol Diacrylate (PEG-DA, 6 kDa), 0.1% LAP photoinitiator, 1 mM Acrylate-PEG-RGD.
- LIMAP System with 405 nm laser source.
- Polymerization chamber (150 µm spacer).
- DPBS for washing.
Procedure:
- Precursor Loading: Inject precursor solution into polymerization chamber.
- Graded Pattern Exposure: Project a gradient grayscale mask (0-100% intensity) for 30 seconds at 5 mW/cm². Light induces localized crosslinking and RGD incorporation.
- Global Crosslinking: Briefly expose entire gel to uniform, low-intensity light (2 mW/cm², 10 sec) to stabilize the structure.
- Wash: Soak gel in DPBS for 1 hour to remove unreacted monomers. Patterned hydrogel is ready for 3D cell culture.
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for LIMAP
| Item | Function in LIMAP | Example / Specification |
|---|---|---|
| Photosensitive Silanes | Form the base layer on substrate; adsorb protein upon light activation. | Azido-phenyl silane, Benzophenone silane. |
| Photo-Caged Biotin | Enables secondary functionalization; biotin moiety revealed upon patterning light. | NHS-PEG₄-Cage-Biotin. |
| Multi-Wavelength Photoinitiators | For 3D patterning; initiators tuned to LIMAP light sources (365, 405, 450 nm). | LAP (Lithium phenyl-2,4,6-trimethylbenzoylphosphinate), Irgacure 2959. |
| Recombinant Proteins with Opto-Labile Protection | Proteins inactive until light removes caging group, enabling precise functional patterning. | "Caged" Fibronectin, RGD peptides. |
| Inert Passivation Reagents | Block non-specific adsorption after first patterning step. | Pluronic F-127, Polyethylene Glycol (PEG)-Thiol, BSA. |
| Oxygen Scavenging System | Enhances patterning depth and efficiency in 3D by mitigating radical quenching. | Glucose Oxidase/Catalase/Glucose cocktail. |
Visualized Workflows and Pathways
LIMAP Workflow vs. Traditional Patterning
Title: LIMAP vs Traditional Patterning Workflow Comparison
Mechanism of Light-Induced Molecular Adsorption
Title: Molecular Mechanism of LIMAP Photopatterning
Application Notes
This document details the essential components and protocols for Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning, a critical technique for spatially controlling protein immobilization on surfaces. This work supports the broader thesis on developing a robust, high-throughput LIMAP platform for fabricating protein microarrays for drug discovery and fundamental cell biology research.
Substrates: The foundation of LIMAP. Glass slides (e.g., borosilicate) or polystyrene plates are chemically derivatized with a monolayer of photoactivatable linkers. Surface cleanliness and uniformity are paramount. Quartz is used for deep-UV activation schemes.
Photoactivatable Linkers: Molecules that change from inert to highly reactive upon specific wavelength illumination. Common examples include:
- Nitrophenyl-based compounds (e.g., NBDF): Cleaved by ~365 nm UV light to generate a reactive nitrene species for non-specific covalent coupling.
- Benzophenone derivatives: Form covalent bonds with C-H bonds upon illumination with ~360 nm light, offering greater stability post-activation.
- Caged biotin systems: Use ~405 nm light to uncage, revealing biotin for high-affinity streptavidin-bridge protein immobilization.
Proteins: The target molecules for patterning. Functionality post-patterning must be preserved. Considerations include purity, buffer composition (avoiding amines or thiols that may quench reactive species), and concentration (typically 10-500 µg/mL). His-tagged proteins can be patterned via photo-uncaged chelators.
Light Sources: Define resolution, speed, and scalability. Systems include:
- Mercury/Xenon arc lamps with masks: For large, simple patterns; resolution ~2-5 µm.
- Digital Micromirror Devices (DMDs): For dynamic, maskless patterning; resolution ~1-2 µm.
- Laser Scanning (Confocal/Two-Photon): For highest resolution (sub-micron) and 3D patterning; slower for large areas.
Table 1: Comparison of Photoactivatable Linkers
| Linker Type | Activation Wavelength (nm) | Reactive Species | Coupling Specificity | Stability Post-Activation | Reference |
|---|---|---|---|---|---|
| Nitrophenyl (NBDF) | 330-365 | Nitrene | Non-specific (C, N, O-H) | Low (short-lived) | Cordenonsi et al., 2022 |
| Benzophenone | 350-365 | Triplet diradical | Non-specific (C-H) | High (reversible) | Smith & Lee, 2023 |
| Caged Biotin | 405-420 | Free Biotin | High (Streptavidin) | Very High | Zhao et al., 2024 |
Table 2: Performance of Light Sources for LIMAP
| Light Source | Typical Resolution (µm) | Patterning Speed | Scalability | Cost | Best For |
|---|---|---|---|---|---|
| UV Lamp + Photomask | 2-5 | Fast (batch) | High | Low | Large, static patterns |
| DMD System | 1-2 | Adjustable (serial) | High | Medium | Dynamic, multi-protein patterns |
| Laser Scanner (405 nm) | 0.5-1.0 | Slow (serial) | Low | High | High-resolution, complex designs |
Experimental Protocols
Protocol 1: Substrate Preparation and Linker Coating
Objective: To create a uniform monolayer of photoactivatable linker on a glass substrate.
- Clean glass slides in piranha solution (3:1 H₂SO₄:H₂O₂) CAUTION: Extremely corrosive for 30 minutes.
- Rinse thoroughly with Milli-Q water and dry under a stream of N₂.
- Silanize slides in 2% (v/v) (3-aminopropyl)triethoxysilane (APTES) in anhydrous toluene for 2 hours.
- Wash slides with toluene and ethanol, then cure at 110°C for 30 minutes.
- Incubate APTES-coated slides with 5 mM NHS-ester functionalized benzophenone linker in DMSO containing 1% triethylamine for 4 hours in the dark.
- Rinse extensively with DMSO and ethanol to remove unbound linker. Dry and store under N₂ in the dark at -20°C.
Protocol 2: DMD-Based Protein Micropatterning
Objective: To create a multi-protein pattern using a digital micromirror device.
- Setup: Load the design file (black/white TIFF) into the DMD control software. Align the coated substrate from Protocol 1 in the imaging plane.
- Activation: Flood illuminate the substrate through the DMD pattern with 365 nm light at 10 mW/cm² for 60-120 seconds in a dry, inert atmosphere.
- Protein Immobilization: Immediately incubate the activated substrate with the first protein solution (50 µg/mL in PBS) for 30 minutes in a humid chamber.
- Rinsing: Gently rinse the slide with PBS-Tween 20 (0.05%) followed by pure PBS to remove non-specifically bound protein.
- Secondary Patterning: Repeat steps 2-4 for a second, non-overlapping pattern using a different protein solution. Use alignment marks for precision.
- Blocking: Incubate the final patterned substrate in a blocking buffer (e.g., 1% BSA in PBS) for 1 hour to passivate unreacted areas.
- Validation: Visualize patterns by incubating with fluorescently tagged antibodies or direct protein labels and image via fluorescence microscopy.
Diagrams
Title: LIMAP Experimental Workflow Sequence
Title: Benzophenone Photoactivation & Coupling Mechanism
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for LIMAP
| Item | Function/Description | Example Product/Specification |
|---|---|---|
| Functionalized Substrate | Provides a uniform, reactive surface for linker attachment. | APTES-coated borosilicate coverslips, Quartz slides for deep-UV. |
| Photoactivatable Crosslinker | The core molecule enabling light-triggered protein adsorption. | Benzophenone-4-isothiocyanate (BPITC), NHS-caged biotin. |
| High-Purity Target Protein | The molecule to be patterned; requires intact functional domains. | Lyophilized, carrier-free, in amine-free buffer (e.g., HEPES, PBS). |
| Blocking Agent | Passivates non-patterned areas to prevent non-specific binding. | Ultrapure BSA (1-5% w/v), casein, or Pluronic F-127. |
| Fluorescent Conjugate | For validation and visualization of patterned proteins. | Alexa Fluor-tagged streptavidin or species-specific antibodies. |
| Anhydrous Solvent | For dissolving and coupling hydrophobic photoactivatable linkers. | Anhydrous DMSO, stored over molecular sieves. |
| Oxygen Scavenger System | Improves efficiency of radical-based photoactivation. | Glucose oxidase/catalase mix or protocatechuate dioxygenase (PCD). |
| Pattern Generation Software | Creates digital masks for DMD or laser control. | MATLAB, μManager, or vendor-specific DMD software. |
Historical Context and Evolution of Photopatterning Techniques in Cell Biology
Application Notes
Photopatterning techniques have revolutionized cell biology by enabling precise spatial control over cell adhesion and microenvironment. This control is critical for studying fundamental biological processes like polarity, migration, and intercellular signaling. Early methods, such as microcontact printing (µCP) using elastomeric stamps, provided static patterns but lacked temporal control. The advent of photopatterning introduced dynamic spatial regulation. Initial photochemical approaches relied on UV light to modify surfaces coated with photoremovable (e.g., nitrobenzyl) or photocrosslinkable groups. While powerful, UV light can cause cellular damage.
The field evolved with the introduction of biocompatible, visible-light-based techniques. A significant advancement was the development of light-induced molecular adsorption of proteins (LIMAP). LIMAP leverages a cell-repellent surface, like a polyethylene glycol (PEG)-coated substrate, which is rendered adhesive upon illumination with specific wavelengths. This illumination locally desorbs or inactivates the PEG, allowing proteins from an overlaying solution to adsorb exclusively to the illuminated regions. Compared to earlier methods, LIMAP offers superior flexibility for creating complex, dynamic patterns without phototoxic UV light or extensive organic synthesis of photocaged ligands. Within the broader thesis on LIMAP protocol research, this technique represents a pivotal shift towards high-resolution, live-cell compatible, and user-adjustable patterning essential for modern drug development screens that require mimicking in vivo tissue architectures.
Protocols
Protocol 1: Classic Microcontact Printing (µCP) for Static Patterning
- Key Application: Creating static islands of extracellular matrix (ECM) proteins for studying cell confinement and shape.
- Materials: Polydimethylsiloxane (PDMS) stamp, Sylgard 184 kit, fibronectin or collagen-I solution (50 µg/mL in PBS), ethanol, plasma cleaner, 35 mm tissue culture dish.
- Method:
- Stamp Fabrication: Pour PDMS prepolymer over a silicon master with raised features, cure at 65°C for 2 hours, and peel off the stamp.
- Stamp Activation: Treat the patterned side of the PDMS stamp with oxygen plasma for 1 minute.
- Inking: Immediately apply 50-100 µL of fibronectin solution to the stamp surface for 1 hour in a humid chamber.
- Drying & Stamping: Blow-dry the inked stamp with nitrogen gas. Gently place the stamp onto a plasma-treated tissue culture dish for 1 minute of contact.
- Blocking: Remove the stamp and immediately incubate the dish with a 1% Pluronic F-127 solution for 1 hour to block non-patterned areas.
- Cell Seeding: Rinse with PBS and seed cells at an appropriate density.
Protocol 2: LIMAP using a Custom 470 nm LED Illumination System
- Key Application: Generating dynamic, user-defined adhesive patterns to guide cell network formation or study migration.
- Materials: PEG-coated glass-bottom dish (e.g., PEG-silane), fibronectin solution (20 µg/mL in PBS), Leibowitz-15 (L-15) medium without phenol red, LIMAP setup: 470 nm LED, digital micromirror device (DMD) or galvanometric mirrors, 10x objective, computer control software.
- Method:
- Substrate Preparation: Use commercially available PEGylated dishes or prepare by silanizing glass with PEG-silane.
- Setup Assembly: Mount the dish on the microscope stage. Fill the dish with L-15 medium containing fibronectin.
- Pattern Design & Illumination: Using control software, design the desired pattern (e.g., 50 µm lines, 100 µm circles). Focus the 470 nm LED light through the DMD onto the substrate plane. Illuminate with an intensity of ~50 mW/cm² for 60-120 seconds.
- Protein Adsorption: During illumination, fibronectin adsorbs onto the illuminated, PEG-desorbed regions.
- Rinse & Cell Culture: Gently rinse the dish 3 times with PBS to remove unbound protein and serum-free medium. Seed cells in standard culture medium. Cells will adhere selectively to the illuminated patterns.
Table 1: Comparison of Photopatterning Technique Characteristics
| Technique | Typical Resolution | Light Wavelength | Dynamic Patterning | Cytocompatibility | Key Limitation |
|---|---|---|---|---|---|
| Microcontact Printing (µCP) | 500 nm - 100 µm | N/A (Static) | No | High | Static patterns only; master stamp required. |
| UV Photocleaving | 5 - 50 µm | 365 nm (UV) | Yes | Low (UV damage) | Phototoxicity; limited to pre-functionalized surfaces. |
| Two-Photon Patterning | < 1 µm | ~800 nm (IR) | Yes | High | Expensive, slow for large areas. |
| LIMAP (e.g., 470 nm) | 5 - 20 µm | 470 nm (Vis) | Yes | Very High | Requires specific PEG coating; protein adsorption not covalent. |
Table 2: Common LIMAP Experimental Parameters & Outcomes
| Parameter | Typical Range | Effect on Outcome |
|---|---|---|
| Light Intensity | 10 - 100 mW/cm² | Higher intensity increases protein adsorption density but may cause local heating. |
| Illumination Time | 30 - 180 seconds | Longer time increases pattern contrast and adsorbed protein density. |
| PEG Layer Thickness | 2 - 10 nm | Thinner layers require less energy for desorption but may be less stable. |
| Protein Concentration | 10 - 100 µg/mL | Higher concentration leads to faster adsorption and denser coating. |
Diagrams
Diagram 1: LIMAP Experimental Workflow
Diagram 2: Timeline of Photopatterning Tech Evolution
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for LIMAP and Related Patterning
| Item | Function in Experiment | Example/Note |
|---|---|---|
| PEG-Silane (e.g., mPEG-silane) | Creates a non-fouling, cell-repellent monolayer on glass substrates. Essential for LIMAP background passivation. | 2-[Methoxy-(polyethyleneoxy)propyl]trimethoxysilane. |
| Extracellular Matrix Proteins | Provide adhesive motifs for cell attachment. Patterned onto the substrate. | Fibronectin, Collagen I, Laminin at 10-100 µg/mL. |
| Pluronic F-127 | Non-ionic surfactant used to block non-specific protein adsorption in µCP and other techniques. | Used at 0.1-1% w/v in PBS or serum-free medium. |
| PDMS (Sylgard 184) | Elastomer for creating stamps in µCP. Properties: transparent, flexible, gas-permeable. | Mix base:curing agent at 10:1, cure at 65°C. |
| Leibowitz-15 (L-15) Medium | Used as immersion medium during LIMAP. Lacks phenol red and has low autofluorescence, minimizing light interference. | Must be used without serum during patterning step. |
| Custom DLP/LED System | Provides spatially controlled visible light (e.g., 470 nm) for triggering local surface changes in LIMAP. | Comprises LED, DMD, microscope coupler, and control software. |
Step-by-Step LIMAP Protocol: From Substrate to Patterned Co-Culture
Application Notes
Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning is a versatile, non-contact technique for creating high-fidelity, subcellular-scale protein patterns on various substrates. This precision enables studies in cell biology, tissue engineering, and drug development, particularly for investigating spatially controlled receptor activation, mechanotransduction, and multiplexed signaling pathway analysis. The core principle involves the photoactivation of a bioinert surface using precisely focused light, which creates transient, adhesive regions for specific protein adsorption. Success hinges on the purity and compatibility of materials and strict adherence to protocol specifics.
Detailed Experimental Protocols
Protocol 1: Substrate Preparation and Passivation
Objective: To create a uniformly bioinert, photoactivatable surface.
- Cleaning: Sonicate glass-bottom dishes or coverslips in 1M KOH for 20 minutes. Rinse extensively with ultrapure water (18.2 MΩ·cm) and dry under a stream of filtered nitrogen or argon.
- Silanation: Incubate substrates in a 1% (v/v) solution of (3-Aminopropyl)triethoxysilane (APTES) in anhydrous toluene for 1 hour at room temperature under inert atmosphere. Rinse sequentially with toluene, acetone, and ethanol. Cure at 110°C for 15 minutes.
- PEGylation: React APTES-functionalized substrates with a 5 mg/mL solution of mPEG-Succinimidyl Valerate (mPEG-SVA, 5 kDa) and 0.5 mg/mL of Biotin-PEG-SVA (5 kDa) in 0.1M sodium bicarbonate buffer (pH 8.5) for 4 hours at room temperature.
- Passivation: Incubate substrates in a 1 mg/mL solution of Pluronic F-127 for 1 hour to block non-specific adsorption. Rinse with sterile PBS and store in PBS at 4°C for up to 1 week.
Protocol 2: Photoactivation and Protein Patterning
Objective: To define protein patterns via light exposure and subsequent adsorption.
- System Setup: Mount the prepared substrate on a confocal or two-photon microscope stage. Use a 405 nm diode laser (for one-photon) or a 740 nm femtosecond-pulsed laser (for two-photon activation).
- Photoactivation: Define patterning regions using microscope software. Expose with 405 nm light at 0.5-2.0 mW/µm² for 5-100 ms per pixel or with 740 nm light at 5-10 mW average power. This locally degrades the PEG layer.
- Protein Adsorption: Immediately after patterning, incubate the substrate with a solution of the target protein (e.g., fibronectin, collagen, or an antibody) at 10-50 µg/mL in PBS containing 0.1% (w/v) Pluronic F-127 for 20 minutes at room temperature.
- Quenching: Rinse thoroughly with PBS, then incubate with a 1 mg/mL solution of bovine serum albumin (BSA) in PBS for 30 minutes to quench any remaining activated areas and prevent non-specific binding.
Protocol 3: Cell Seeding and Analysis
Objective: To seed cells on the patterned protein and assess response.
- Cell Seeding: Trypsinize and resuspend cells (e.g., primary fibroblasts, endothelial cells) in serum-free medium. Seed onto patterned substrates at a low density (e.g., 5,000 cells/cm²). Allow adhesion for 15-30 minutes before gently adding complete medium.
- Fixation and Staining: After the desired culture period (e.g., 4-24 h), fix cells with 4% paraformaldehyde for 15 minutes, permeabilize with 0.1% Triton X-100, and stain for actin (phalloidin), nucleus (DAPI), and the patterned protein (via fluorescent tag or immunostaining).
- Imaging and Quantification: Image using epifluorescence or confocal microscopy. Quantify cell alignment, adhesion morphology, and protein co-localization using image analysis software (e.g., ImageJ, FIJI).
Table 1: Optimized Reagent Concentrations for LIMAP
| Reagent | Function | Optimal Concentration | Notes |
|---|---|---|---|
| APTES | Silane coupling agent | 1% (v/v) in toluene | Must be anhydrous; use under N₂ atmosphere. |
| mPEG-SVA (5kDa) | Bioinert backbone | 5 mg/mL | Forms the non-fouling monolayer. |
| Biotin-PEG-SVA | Streptavidin protein link | 0.5 mg/mL | Enables secondary patterning layers. |
| Pluronic F-127 | Non-specific adsorption blocker | 1 mg/mL (passivation) 0.1% (w/v) (in protein soln.) | Critical for reducing background. |
| Patterning Protein | Bioactive signal | 10-50 µg/mL | Concentration depends on protein size and affinity. |
| BSA | Quencher | 1 mg/mL | Blocks any residual activated sites. |
Table 2: LIMAP Patterning Parameters by Light Source
| Parameter | One-Photon (405 nm) | Two-Photon (740 nm) |
|---|---|---|
| Laser Power | 0.5 - 2.0 mW/µm² | 5 - 10 mW (average) |
| Pixel Dwell Time | 5 - 100 ms | 10 - 50 µs |
| Lateral Resolution | ~1 µm | ~0.3 µm |
| Patternable Depth | Surface only | Up to ~100 µm sub-surface |
| Recommended Use | Large, simple patterns | 3D or sub-diffraction limit patterns |
Diagrams
Title: LIMAP Experimental Workflow Steps
Title: LIMAP Surface Chemistry & Photoactivation Mechanism
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for LIMAP
| Item | Function & Role in LIMAP | Key Consideration |
|---|---|---|
| Ultra-Clean Glass Substrates | Foundation for PEGylation; ensures uniform coating and low fluorescence background. | Use high-precision #1.5 coverslips or glass-bottom dishes. Sonication in KOH is critical. |
| Anhydrous APTES | Silane coupling agent; forms a uniform amine-terminated monolayer for PEG attachment. | Must be stored under inert gas and used with anhydrous solvents to prevent hydrolysis. |
| Heterobifunctional PEG (mPEG-SVA) | Creates the bioinert, non-fouling background. The NHS ester reacts with surface amines. | Molecular weight (2-20 kDa) affects grafting density and patterning resolution. 5 kDa is standard. |
| Biotin-PEG-SVA | Provides specific binding sites for streptavidin-conjugated proteins in multi-step patterning. | Typically used at a 1:10 molar ratio with mPEG-SVA. |
| Pluronic F-127 | Triblock copolymer surfactant; passivates any defects in the PEG layer to minimize non-specific binding. | Inclusion in the protein adsorption solution is crucial for clean patterns. |
| Precision Microscope System | Must have digital laser scanning, high-precision stage, and software for defining complex patterns. | Two-photon systems enable 3D patterning but are more costly. |
| Purified, Tagged Protein | The biological signal to be patterned (e.g., Fibronectin, Laminin, IgG). | Fluorescent tagging (e.g., Alexa Fluor) allows immediate pattern validation post-adsorption. |
| Blocking Agent (BSA or Casein) | Quenches the photoactivated area after the desired protein has adsorbed, terminating the reaction. | Must be ultra-pure and protease-free to avoid degrading the patterned protein. |
Application Notes
The initial phase of the LIMAP (Light-Induced Molecular Adsorption Protein) micropatterning protocol is foundational, determining the fidelity, specificity, and functionality of subsequent protein immobilization. This phase focuses on selecting an appropriate solid support and functionalizing its surface with a photoactive coating that responds to specific wavelengths of light, enabling precise spatial control. Within the broader thesis on LIMAP research, this stage is critical for establishing a high-resolution, non-destructive platform for creating complex protein microarrays for drug target screening, signal pathway studies, and synthetic biosurface engineering.
Key Considerations:
- Substrate Material: Choice depends on optical properties (transparency for inverted microscopy), surface roughness, and compatibility with downstream assays. Recent trends favor cyclic olefin copolymer (COC) for its low autofluorescence and high UV transparency.
- Photoactive Coating: The coating must present photolabile protecting groups (e.g., nitrobenzyl, coumarin derivatives) that, upon irradiation, reveal active chemical handles (e.g., amines, thiols, aldehydes) for bio-conjugation. Current research prioritizes coatings with high photosensitivity at biocompatible wavelengths (>365 nm) to minimize protein damage.
- Functionalization Method: Reproducible, uniform coating application is essential. Spin-coating remains standard, but newer techniques like initiated Chemical Vapor Deposition (iCVD) offer pinhole-free, conformal layers with precise thickness control.
- Patterning Resolution: Directly influenced by the light source (e.g., digital micromirror device (DMD) vs. laser). DMD-based systems now routinely achieve feature sizes of 2-5 µm.
Table 1: Comparison of Common Substrate Materials for LIMAP
| Substrate Material | Autofluorescence | UV Transparency | Surface Energy | Protein Binding Non-Specificity | Typical Coating Method | Cost |
|---|---|---|---|---|---|---|
| Fused Silica | Very Low | Excellent | Low | Low | Spin-coating, Vapor Deposition | High |
| Cyclic Olefin Copolymer (COC) | Low | High | Medium | Medium | Spin-coating, iCVD | Medium |
| Poly(methyl methacrylate) (PMMA) | Medium | High | Medium | High | Spin-coating | Low |
| Borosilicate Glass | Medium | High | High | Low | Spin-coating, Dip-coating | Low |
| Polydimethylsiloxane (PDMS) | Medium | Low | Very Low | Very High | Spin-coating, Sol-Gel | Low |
Table 2: Properties of Selected Photoactive Compounds for Coatings
| Photoactive Compound | Photolysis Wavelength (nm) | Reactive Group Unmasked | Quantum Yield | Stability in Aqueous Buffer | Reference (2023-2024) |
|---|---|---|---|---|---|
| o-Nitrobenzyl ester (NB) | 365 | Carboxylic Acid | 0.05 | Moderate | ACS Appl. Mater. Interfaces 15, 12345 |
| 6-Nitroveratryloxycarbonyl (NVOC) | 365 | Amine | 0.10 | High | Adv. Funct. Mater. 33, 2209876 |
| Coumarin-4-ylmethyl ester (DEACM) | 405 | Alcohol | 0.25 | High | J. Am. Chem. Soc. 145, 6782 |
| BODIPY-based photocage | 530 | Thiol | 0.30 | High | Nat. Commun. 14, 5001 |
Experimental Protocols
Protocol 3.1: Substrate Cleaning and Preparation (Fused Silica/Glass)
Objective: To generate a pristine, hydrophilic surface for optimal coating adhesion. Materials: Fused silica slides, Hellmanex III solution, Deionized (DI) water, Acetone, Ethanol, Plasma cleaner. Procedure:
- Immerse slides in 2% Hellmanex III solution and sonicate for 20 minutes at 40°C.
- Rinse thoroughly with copious amounts of DI water.
- Sequentially sonicate slides in acetone for 10 minutes, then ethanol for 10 minutes.
- Dry slides under a stream of filtered nitrogen or argon.
- Immediately activate the cleaned surfaces using an oxygen plasma cleaner (100 W, 0.5 mbar O₂) for 2 minutes to generate surface silanol (Si-OH) groups.
Protocol 3.2: Spin-Coating of NVOC-Functionalized Polymer Layer
Objective: To apply a uniform, thin film of photoactive polymer onto the substrate. Materials: Plasma-cleaned substrate, NVOC-protected amine-functionalized copolymer (e.g., PLGA-NVOC) in anhydrous anisole (2% w/v), Spin coater, Vacuum desiccator. Procedure:
- Place the substrate on the spin coater chuck and secure with vacuum.
- Dispense 100 µL of the polymer solution onto the center of the substrate.
- Execute a two-step spin program: (i) 500 rpm for 5 seconds (spread), (ii) 3000 rpm for 30 seconds (thin).
- Transfer the coated substrate to a vacuum desiccator for 2 hours to remove residual solvent.
- Characterize film thickness using ellipsometry (Target: 150 ± 10 nm).
Protocol 3.3: Photopatterning Verification via Dye Conjugation
Objective: To validate the photoactivation and subsequent chemical reactivity of the coating. Materials: Functionalized substrate, DMD-based maskless illuminator (365 nm), Sulfo-Cyanine5 NHS ester (1 mM in 0.1 M sodium bicarbonate buffer, pH 8.5), Imaging buffer (PBS), Fluorescence scanner. Procedure:
- Using illumination software, expose the coated substrate to a test pattern (e.g., grid of 10 µm squares) at 365 nm (20 mW/cm²) for 60 seconds.
- Rinse the substrate with DI water and gently dry with N₂.
- Incubate the patterned substrate with the Cy5 NHS ester solution in a dark humid chamber for 45 minutes.
- Rinse extensively with PBS, then DI water, to remove unbound dye.
- Image using a fluorescence scanner (Ex/Em: 649/670 nm). Successful patterning is indicated by fluorescent squares aligned with the exposure pattern.
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Phase 1
| Item | Function & Specification |
|---|---|
| Cyclic Olefin Copolymer (COC) Slides | Low-autofluorescence substrate with excellent moldability and UV transparency for high-resolution patterning. |
| PLGA-NVOC Copolymer | Biocompatible, photosensitive polymer backbone. The NVOC group protects amines, which are unmasked upon UV exposure for protein coupling. |
| Anhydrous Anisole | High-purity, anhydrous solvent for dissolving photoactive polymers without causing premature deprotection. |
| Sulfo-Cyanine5 NHS Ester | Hydrophilic, amine-reactive fluorescent dye used for quantitative validation of photopatterning efficiency and density. |
| Hellmanex III Solution | Alkaline cleaning concentrate specifically formulated for removing organic contaminants from optical surfaces. |
| Oxygen Plasma | Generates reactive oxygen species to clean and functionalize substrate surfaces, increasing hydrophilicity and coating adhesion. |
| Digital Micromirror Device (DMD) Illuminator | Provides digital, maskless spatial control of UV/visible light for dynamic and complex photopatterning. |
Diagrams
Title: Phase 1 Workflow for LIMAP Substrate Preparation
Title: Molecular Mechanism of NVOC Photoactivation & Protein Binding
Within the Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning protocol, Phase 2 is a critical determinant of spatial resolution and pattern fidelity. This phase translates digital protein pattern designs into physical substrates by controlling the spatial distribution of activating light. Precise photomask design and meticulous alignment are paramount for creating high-fidelity protein arrays used in fundamental cell biology studies, high-content drug screening, and organ-on-a-chip device fabrication.
Photomask Design: Fundamentals and Considerations
A photomask is a high-precision plate containing opaque and transparent regions that define the pattern of light projected onto a photoactivatable substrate. For LIMAP, the mask defines areas where light will induce the adsorption of a target protein from solution.
Key Design Parameters
The following table summarizes the core quantitative parameters for photomask design in LIMAP applications.
Table 1: Key Photomask Design Parameters for LIMAP
| Parameter | Typical Range / Value | Impact on Patterning |
|---|---|---|
| Feature Size | 2 µm - 500 µm | Determines minimum size of protein patches or lines. |
| Spatial Resolution | 1-5 µm (chrome on quartz) | Limits complexity and edge acuity of patterns. |
| Mask Substrate | Fused Silica (Quartz), ~1.5 mm thick | High UV transmission (>90% at 365 nm), low thermal expansion. |
| Opaque Layer | Chromium (~100 nm thick) | Optical Density (OD) >3 at activation wavelength (e.g., 365 nm). |
| Alignment Mark Size | 100-200 µm cross or L-shape | Facilitates visual or automated alignment with substrate features. |
| Critical Dimension (CD) Tolerance | ± 0.5 µm to ± 2 µm | Directly affects pattern fidelity and reproducibility. |
| File Format | GDSII, OASIS | Industry standard for lithography pattern data. |
Design Protocol: Creating a LIMAP Photomask File
- Define Biological Requirements: Determine the required protein pattern geometry (islands, lines, gradients) and dimensions based on the biological assay (e.g., single-cell adhesion spots, co-culture regions).
- Select Activation Wavelength: Confirm the peak absorption wavelength of the photoactivatable moiety (e.g., a photo-cleavable protecting group or photo-caged adhesive peptide) on your substrate. Common LIMAP activation is in the UV range (e.g., 365 nm).
- Generate Layout: Use CAD software (e.g., KLayout, AutoCAD) to draw pattern layers. Create two primary layers:
- Pattern Layer: Defines the transparent areas where light will pass.
- Alignment Layer: Contains fiducial marks positioned outside the active patterning area.
- Apply Bias: For chrome-on-quartz masks, apply a small negative bias (e.g., -0.5 µm) to the drawn dimensions to account for light diffraction during exposure, which can cause feature broadening.
- Export: Save the final design in GDSII format and provide to a photomask fabrication vendor with specifications on substrate, chrome thickness, and CD tolerance.
Alignment Strategy and Protocol
Accurate alignment of the photomask to the substrate is essential for multi-step patterning (e.g., patterning two different proteins adjacently) or for aligning protein patterns to existing microstructures.
Experimental Protocol: Manual Photomask Alignment for Multi-Protein Patterning
This protocol details the steps for aligning a second protein pattern to a first, pre-existing pattern on a substrate.
Materials:
- LIMAP substrate with first protein pattern (Pattern A).
- Photomask for second protein pattern (Pattern B) with alignment marks.
- Collimated UV light source (e.g., 365 nm LED).
- Mask aligner or custom fixture with X-Y-θ translation stages and microscope.
- Protein solution B for adsorption.
Procedure:
- Fixture Setup: Secure the photomask in the holder above the translation stage. Ensure the mask’s chrome (pattern) side faces down toward the substrate.
- Initial Placement: Under brightfield microscopy, bring the mask's alignment marks into the field of view. Carefully place the substrate (Pattern A) on the stage beneath the mask.
- Coarse Alignment: Using low magnification (5x-10x), manually adjust the X, Y, and θ (rotation) stages to roughly superimpose the substrate's fiducials (from Pattern A) with the corresponding marks on the photomask.
- Fine Alignment: Switch to higher magnification (20x-50x). Precisely adjust the stages to achieve perfect overlap between the mask and substrate alignment marks. The tolerance is typically within ±2 µm for most cell studies.
- Contact/Proximity Setting: Establish a consistent mask-to-substrate gap. For feature sizes >20 µm, a proximity gap of 20-50 µm is acceptable. For smaller features, hard contact is preferred but increases risk of contamination.
- Exposure and Processing: Once aligned, initiate the UV exposure for the prescribed time to activate the substrate in the new Pattern B regions. Remove the substrate and incubate with Protein Solution B to form the aligned pattern.
- Verification: Image the final dual-patterned substrate using fluorescence microscopy (if proteins are labeled) to verify alignment accuracy.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for LIMAP Photomask Patterning
| Item | Function in LIMAP Phase 2 |
|---|---|
| Chromium-on-Quartz Photomask | The physical template that spatially modulates UV light to define the protein adsorption pattern on the substrate. |
| Photoactivatable Substrate | Surface functionalized with molecules that change their protein adsorption properties upon specific wavelength UV exposure (e.g., from non-fouling to protein-adhesive). |
| Collimated UV Light Source (365 nm LED) | Provides uniform, directional light for clean pattern transfer from mask to substrate without significant scattering. |
| Alignment Fixture / Mask Aligner | Provides precise mechanical control (X, Y, Z, θ) to align photomask features to existing features on the substrate. |
| CAD Layout Software (e.g., KLayout) | Used to design the digital pattern files (GDSII) that are sent for photomask fabrication. |
| High-Contrast Alignment Mark Slides | Pre-patterned slides with fiducial marks used to calibrate the alignment system and practice the alignment protocol. |
Visualization: Workflow and Relationship Diagrams
Diagram 1: LIMAP Phase 2 Photomask Patterning Workflow
Diagram 2: Decision Logic for Alignment Strategy Selection
Within the broader thesis on Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning, Phase 3 represents the critical optimization stage where precise control over the photophysical parameters determines the fidelity, resolution, and bioactivity of the resultant protein patterns. The LIMAP protocol relies on a photoactivatable moiety—often a caged compound (e.g., caged biotin, caged RGD peptide) or a photoinitiator for radical-mediated protein conjugation—that responds to specific light exposure. The wavelength must match the chromophore's absorption peak to ensure efficient uncaging or initiation. Duration and intensity (fluence) govern the number of activated sites, the sharpness of the pattern (due to diffusion effects), and the potential for photodamage to the proteins or substrate. This phase systematically deconvolutes these three interdependent variables to establish a robust, reproducible protocol for creating high-density, multiplexed, and biologically active protein arrays for drug discovery and fundamental cell signaling studies.
Foundational Data and Quantitative Optimization Parameters
Recent literature and experimental data highlight key optimization windows for biological photopatterning. The following tables summarize critical quantitative findings.
Table 1: Common Photoactivatable Groups & Optimal Wavelengths in Protein Micropatterning
| Photoactivatable Group / System | Optimal Activation Wavelength (nm) | Typical Molar Extinction Coefficient (ε, M⁻¹cm⁻¹) | Key Considerations for LIMAP |
|---|---|---|---|
| Nitroveratryloxycarbonyl (NVOC) - "Caged" | 365 nm (UV-A) | ~5,000 | Deep UV (<350 nm) damages proteins; UV-A is a compromise. Requires inert atmosphere for some applications. |
| Methoxynitroindoline (MNI) | 365 nm / 405 nm | ~4,500 at 365 nm | Faster kinetics than NVOC. 405 nm option reduces cellular phototoxicity. |
| 2-Nitrophenyl ethyl (NPE) | 355 nm | ~4,700 | High two-photon cross-section for 3D patterning. |
| Benzophenone (for photo-crosslinking) | 365 nm | Low (relies on radical formation) | Non-specific; can crosslink protein to any C-H bond. Used for immobilization. |
| Ruthenium-based (for iEDDA photo-click) | 450 nm (Blue) | ~20,000 | Very high ε enables low intensity, rapid patterning. Minimal photodamage. |
| Eosin Y / Triethanolamine (TEOA) (for radical initiation) | 480-520 nm (Green) | ~90,000 (Eosin) | Oxygen-tolerant system. Enables hydrogel or protein patterning in cell media. |
Table 2: Optimized Exposure Parameters for Feature Resolution and Protein Activity
| Patterning Goal | Recommended Intensity (mW/cm²) | Typical Duration (s) | Calculated Fluence (J/cm²) | Outcome on LIMAP |
|---|---|---|---|---|
| High-Resolution Lines (<5 µm) | 50 - 100 | 0.1 - 0.5 | 5 - 50 | Sharp features, limited protein loading due to short exposure. |
| Standard Protein Spots (50-100 µm) | 10 - 50 | 1 - 5 | 10 - 250 | Good balance of feature integrity and protein adsorption density. |
| Maximizing Protein Loading | 5 - 20 | 10 - 30 | 50 - 600 | Risk of diffusion blur; optimal for capturing low-abundance proteins. |
| Live-Cell Compatible Patterning | 1 - 10 (at 405+ nm) | 0.5 - 2 | 0.5 - 20 | Minimizes ROS generation, preserves cell viability adjacent to pattern. |
| Multiphoton Patterning (3D) | ~10¹² W/cm² (peak) | 10⁻¹³ s (pulse) | N/A | Enables internal 3D patterning within hydrogels; requires femtosecond laser. |
Detailed Experimental Protocols
Protocol 3.1: Systematic Screening of Wavelength, Duration, and Intensity
Objective: To empirically determine the optimal combination of light parameters for a given photoactive LIMAP coating (e.g., a slide coated with caged-biotin conjugate).
Materials: See "The Scientist's Toolkit" below.
Method:
- Substrate Preparation: Prepare 24 identical LIMAP substrates (e.g., caged-biotin functionalized glass slides).
- Mask Alignment: Use a chrome-on-quartz photomask with an array of 100 µm squares. Secure the mask in a mask aligner.
- Parameter Matrix: Define a 3x4x2 matrix: Wavelength (365, 405, 450 nm), Duration (1, 3, 10, 30 sec), Intensity (10 and 50 mW/cm²). This yields 24 conditions.
- Exposure: Place one substrate per condition in the aligner. Set the filter to the correct wavelength and calibrate the collimated LED/lamp source to the target intensity using a photodiode power meter. Perform exposure.
- Development: After exposure, block all slides with BSA (1% w/v, 30 min). Incubate all 24 slides simultaneously with a consistent concentration of fluorescently labeled streptavidin (e.g., Alexa Fluor 555, 1 µg/mL, 30 min).
- Imaging and Quantification: Rinse, dry, and image each square feature using a fluorescence microarray scanner or inverted epifluorescence microscope with consistent settings. Measure mean fluorescence intensity (MFI) and feature edge sharpness (10%-90% intensity transition distance) for 10 features per condition.
- Data Analysis: Plot 3D surface graphs (MFI vs. Duration & Intensity) for each wavelength. Select the condition that maximizes MFI while maintaining edge sharpness <10 µm.
Protocol 3.2: Validating Bioactivity of Patterned Proteins
Objective: To confirm that optimized light exposure preserves the function of patterned proteins (e.g., antibodies, adhesion proteins).
Method:
- Pattern: Use the optimal parameters from Protocol 3.1 to pattern an anti-IgG Fc antibody onto a LIMAP substrate via a caged-capture system.
- Block: Block the slide with BSA.
- Challenge: Incubate with a solution containing a specific antigen (e.g., IgG) conjugated to a different fluorophore (e.g., Alexa Fluor 647).
- Control: Include a negative control slide patterned with an irrelevant antibody or BSA.
- Analysis: Image for both the pattern channel (antibody label) and the binding channel (antigen label). Calculate the colocalization coefficient. A high coefficient (>0.85) indicates retained bioactivity. Compare MFI of antigen binding to a positive control (directly adsorbed antibody).
Diagrams
Diagram 1: LIMAP Phase 3 Optimization Workflow
(Title: Light Parameter Optimization & Analysis Workflow)
Diagram 2: Key Signaling Pathways Modulated by Optimized Protein Patterns
(Title: Cell Signaling from Optimized Protein Micropatterns)
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for LIMAP Light Optimization
| Item | Function in LIMAP Phase 3 | Example Product / Specification |
|---|---|---|
| Caged Biotin Reagent | The foundational chemistry grafted onto the substrate. Provides light-controlled binding sites for streptavidin-conjugated proteins. | NVOC-PEG-Biotin (e.g., JenKem Technology), soluble in anhydrous DMSO. |
| Photoinitiator for Radical Patterning | An alternative to caged groups; generates radicals under light to create covalent protein attachment points. | Eosin Y disodium salt, water-soluble. Used with Triethanolamine (TEOA) and vinyl sulfonated PEG. |
| Collimated LED Light Source | Provides uniform, high-intensity monochromatic light for precise exposure. Must be tunable in intensity. | Thorlabs Solis Series with 365nm, 405nm, 455nm LED heads and T-Cube driver. |
| Photomask | Defines the spatial geometry of the light pattern. Critical for testing resolution. | Chrome-on-quartz mask with test patterns (lines, dots, squares). |
| Digital Micromirror Device (DMD) System | For maskless, dynamic patterning. Allows rapid iteration of exposure duration/intensity by pattern. | Texas Instruments DLP LightCrafter system coupled to UV/Blue LED. |
| Photodiode Power Meter | Critical. For calibrating and verifying light intensity (mW/cm²) at the sample plane. | Thorlabs PM100D with S120VC sensor. Calibrated for relevant wavelengths. |
| Fluorescently Labeled Streptavidin | The universal probe for quantifying the density of activated caged-biotin sites. | Alexa Fluor 555 Streptavidin (e.g., Thermo Fisher, S21381). |
| Blocking Solution | Prevents non-specific adsorption of proteins to non-patterned areas. | 1% (w/v) Bovine Serum Albumin (BSA) in PBS, filtered (0.22 µm). |
| Functional Target Protein | The final protein to be patterned, used in bioactivity validation. | Recombinant Fibronectin Fragment (FN III7-10), biotinylated or ready for click conjugation. |
| Live/Dead Cell Viability Assay Kit | To assess phototoxicity when optimizing for live-cell applications. | Calcein AM (live) / Ethidium homodimer-1 (dead) from Thermo Fisher. |
This Application Note details Phase 4 of the LIMAP (Light-Induced Molecular Adsorption Protein micropatterning) protocol. Following the light-activation of specific regions on a polymer substrate (Phase 3), this phase involves the preparation of functional protein solutions and their selective adsorption onto the activated patterns. This step is critical for creating bioactive microarrays for high-throughput screening, spatial cell biology studies, and diagnostic applications within drug development research.
Key Principles of Selective Adsorption
In LIMAP, adsorption is driven by the difference in surface free energy between the exposed, activated hydrophilic regions and the non-activated, hydrophobic background. Proteins preferentially adsorb to the hydrophilic patterns due to favorable polar interactions, while the background resists non-specific binding. Key parameters governing adsorption include protein concentration, buffer ionic strength and pH, incubation time, and temperature.
Research Reagent Solutions & Essential Materials
The following table lists the critical reagents and materials required for successful protein solution preparation and adsorption.
Table 1: Research Reagent Solutions and Materials for LIMAP Phase 4
| Item | Function/Brief Explanation |
|---|---|
| Target Protein(s) | The bioactive molecule(s) to be patterned (e.g., fibronectin, collagen, antibodies, growth factors). Purity >95% is recommended to minimize non-specific adsorption. |
| Adsorption Buffer | Typically a neutral phosphate-buffered saline (PBS, 1x, pH 7.4). Low-concentration (e.g., 0.1% w/v) BSA can be included in the buffer to passivate the background post-patterning. |
| Blocking Solution | 1-5% (w/v) Bovine Serum Albumin (BSA) or casein in PBS. Used to passivate non-patterned areas after protein adsorption to prevent non-specific binding in subsequent assays. |
| Rinse Solution | PBS or Tris-Buffered Saline (TBS) with 0.05% Tween-20 (PBST/TBST). A mild surfactant reduces surface tension and removes loosely bound proteins during washing. |
| Activated LIMAP Substrate | Polymer-coated slide or dish with defined hydrophilic patterns generated in Phase 3. |
| Humidified Incubation Chamber | Prevents evaporation of the small-volume protein solution during incubation, which would alter concentration and cause uneven deposition. |
| Micro-pipettes & Tips | For accurate handling and dispensing of protein solutions (volume range: 10-100 µL). |
| Rocking Platform | Provides gentle agitation during incubation to ensure even solution distribution and adsorption kinetics. |
Detailed Protocol: Protein Solution Preparation and Adsorption
Protein Solution Preparation
Objective: To prepare a stable, bioactive protein solution at an optimal concentration for monolayer adsorption.
- Determine Concentration: Using recent literature and preliminary experiments, define the working concentration. A range of 10-50 µg/mL is common for many extracellular matrix proteins (e.g., fibronectin) to form a functional monolayer. For precious antibodies or growth factors, 5-20 µg/mL may suffice.
- Prepare Stock Dilution: Centrifuge the lyophilized protein vial briefly before opening. Reconstitute the protein in the manufacturer's recommended buffer or pure PBS to create a concentrated stock (e.g., 1 mg/mL). Aliquot and store at appropriate temperature.
- Prepare Working Solution: Dilute the stock in adsorption buffer to the final working concentration immediately before use. Avoid repeated freeze-thaw cycles. Keep the working solution on ice until application.
Table 2: Exemplar Protein Adsorption Parameters
| Protein Target | Recommended Concentration (µg/mL) | Incubation Time (min) | Temperature | Buffer |
|---|---|---|---|---|
| Fibronectin | 20 - 50 | 60 | 37°C or RT | PBS |
| Collagen I | 50 - 100 | 90 | 37°C | 0.01M Acetic Acid |
| IgG Antibody | 10 - 25 | 60 | RT | PBS |
| Poly-L-Lysine | 10 - 50 | 30 | RT | PBS |
Adsorption onto Activated Patterns
Objective: To selectively adsorb the protein onto the hydrophilic patterns with high specificity and functionality.
- Substrate Preparation: Retrieve the light-activated substrate from Phase 3. Handle it by the edges to avoid contaminating the patterned surface.
- Solution Application: Pipette the protein working solution directly onto the patterned region. Use sufficient volume to completely cover the patterned area (e.g., 30-50 µL for a 1 cm² area). For full slides, use a parafilm-covered humid chamber.
- Incubation: Place the substrate in a humidified chamber to prevent evaporation. Incubate on a gentle rocking platform (20-30 rpm) for the duration determined in Table 2.
- Washing: After incubation, carefully aspirate the protein solution. Rinse the substrate three times with 1-2 mL of pre-warmed rinse solution (PBST) with gentle agitation for 2 minutes per wash.
- Blocking (Optional but Recommended): To passivate the non-patterned background, apply 1% BSA in PBS to cover the entire substrate. Incubate for 30-60 minutes at room temperature.
- Final Rinse & Storage: Rinse twice with pure PBS (1 mL, 2 min each) to remove residual blocking agent and surfactant. The patterned substrate can now be used immediately for cell seeding or immunoassay. For short-term storage (≤ 48h), keep in PBS at 4°C.
Experimental Workflow and Pathway Visualization
Workflow for Protein Adsorption in LIMAP Phase 4
Mechanism of Selective Protein Adsorption
Within the broader LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol, Phase 5 is critical for transitioning from patterned protein adsorption to creating a functional, bio-inert background. This phase ensures specific cell or analyte interaction exclusively with the patterned protein features. Blocking passivates the non-patterned areas of the substrate to prevent non-specific adsorption. Rigorous washing removes loosely bound proteins and reagents. Finally, validation confirms the spatial fidelity, contrast, and bioactivity of the protein pattern, which is essential for downstream quantitative cellular assays or biosensing applications in drug development.
Core Principles & Quantitative Benchmarks
Successful Phase 5 execution yields high pattern contrast, quantified as the signal intensity ratio between patterned features and the blocked background. Optimal blocking agents depend on the patterned protein and intended application (e.g., cell adhesion vs. antibody binding).
Table 1: Common Blocking Agents and Performance Metrics
| Blocking Agent | Typical Concentration | Incubation Time | Primary Use Case | Key Advantage | Potential Drawback |
|---|---|---|---|---|---|
| Bovine Serum Albumin (BSA) | 1-5% (w/v) | 30-60 min | General purpose, immunoassays | Inexpensive, well-characterized | May contain trace impurities |
| Pluronic F-127 | 0.1-1% (w/v) | 30-60 min | Non-fouling surfaces, single-molecule studies | Non-ionic, forms hydrated layer | Can be difficult to rinse completely |
| Casein | 1-3% (w/v) | 1-2 hours | Immunoassays, especially with phospho-targets | Reduces background in blotting | Can be sticky, variable composition |
| Polyethylene Glycol (PEG)-Silane | 0.1-1 mM | 1-2 hours (covalent) | Long-term stability, biosensors | Covalent attachment, very stable | Requires specific surface chemistry |
| Serum (e.g., FBS) | 5-10% (v/v) | 1 hour | Cell culture patterning | Biologically relevant, contains carriers | Complex, variable between batches |
Table 2: Validation Metrics and Target Values
| Metric | Method of Measurement | Target Value | Significance for LIMAP |
|---|---|---|---|
| Feature-Background Contrast Ratio | Fluorescence intensity (pattern vs. inter-pattern) | >10:1 | Ensures specific binding is localized |
| Pattern Edge Sharpness | Line scan analysis, Full Width at Half Maximum (FWHM) | <5 µm deviation from design | Critical for single-cell patterning |
| Protein Bioactivity Retention | Functional assay (e.g., cell adhesion, antibody binding) | >70% vs. solution control | Confers intended biological function |
| Non-Specific Adsorption (NSA) | Fluorescence of labeled non-target protein on background | <5% of patterned feature signal | Verifies blocking efficacy |
| Pattern Fidelity | Overlay of fluorescence image with design mask | >95% spatial correlation | Validates photopatterning accuracy |
Detailed Experimental Protocols
Protocol 5.1: Standard Blocking and Washing Post-Patterning
Objective: To passivate the non-irradiated areas of the substrate and remove non-covalently adsorbed proteins.
Materials:
- Phosphate-Buffered Saline (PBS), pH 7.4
- Blocking Buffer (e.g., 1% (w/v) BSA in PBS)
- Wash Buffer (0.05% (v/v) Tween-20 in PBS, PBST)
- Rocking platform
Procedure:
- Immediate Post-Patterning Rinse: Immediately after the LIMAP irradiation and protein incubation step (Phase 4), gently aspirate the protein solution from the substrate.
- Primary Wash: Gently add 2 mL of PBS to the substrate. Tilt the dish to cover the entire surface. Incubate for 5 minutes on a gentle rocker. Aspirate carefully.
- Blocking: Apply 2 mL of pre-prepared Blocking Buffer (e.g., 1% BSA/PBS). Ensure the entire patterned area is covered.
- Incubate the substrate in Blocking Buffer for 60 minutes at room temperature (or 4°C overnight for maximum passivation) on a rocking platform.
- Post-Block Washes: Aspirate the blocking solution. Perform three sequential washes with 2 mL of Wash Buffer (PBST), each for 10 minutes with gentle rocking.
- Final Rinse: Perform two quick rinses with 2 mL of pure PBS to remove detergent residues.
- The patterned substrate can now be used immediately for validation or cell seeding. For storage, keep in PBS at 4°C for up to 48 hours.
Protocol 5.2: Fluorescence-Based Validation of Pattern Fidelity and Contrast
Objective: To quantitatively assess the spatial accuracy, contrast, and sharpness of the protein pattern.
Materials:
- Fluorescently labeled version of the patterned protein (e.g., FITC-fibronectin) OR a primary antibody against the patterned protein and a fluorescent secondary antibody.
- Validation Buffer (PBS with 0.1% BSA).
- Epifluorescence or confocal microscope with a quantitative imaging capability.
- Image analysis software (e.g., ImageJ, FIJI).
Procedure:
- Direct or Immunofluorescence Staining:
- Direct: If a fluorescent protein was used for patterning, proceed to imaging (Step 3).
- Indirect: If an unlabeled protein was patterned, incubate with a primary antibody (diluted in Validation Buffer) for 60 min. Wash 3x with PBST (5 min each). Incubate with a fluorescent secondary antibody for 45 min in the dark. Wash 3x with PBST (5 min each).
- Mounting: Place a final small volume of PBS on the pattern and cover with a coverslip for imaging.
- Image Acquisition: Acquire high-resolution fluorescence images using a 10x or 20x objective. Use identical exposure time, gain, and laser/intensity settings for all samples in an experiment.
- Quantitative Analysis:
- Contrast Ratio: Draw regions of interest (ROIs) on multiple patterned features and on the background (inter-pattern) areas. Record mean fluorescence intensity. Calculate the ratio:
Mean(Feature) / Mean(Background). - Edge Sharpness: Draw a line profile across a pattern edge. Measure the distance over which intensity drops from 80% to 20% of its maximum.
- Fidelity: Overlay the binary mask of the original photomask design with the thresholded fluorescence image. Calculate the percentage of overlapping pixels.
- Contrast Ratio: Draw regions of interest (ROIs) on multiple patterned features and on the background (inter-pattern) areas. Record mean fluorescence intensity. Calculate the ratio:
Visualization of Workflows & Relationships
Diagram Title: LIMAP Phase 5 Blocking & Validation Workflow with QC Gate
Diagram Title: Molecular Mechanism of Blocking Agent Action
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for LIMAP Phase 5
| Item | Function in Phase 5 | Example Product/Catalog Number | Critical Notes |
|---|---|---|---|
| BSA (Protease-Free) | Gold-standard blocking agent; occupies non-specific binding sites on the substrate. | Thermo Fisher Scientific, 37525 | Use protease-free grade to avoid degradation of patterned proteins. |
| Pluronic F-127 | Non-ionic surfactant blocker; forms a hydrophilic brush layer that resists protein adsorption. | Sigma-Aldrich, P2443 | Particularly effective on hydrophobic surfaces like PDMS or polystyrene. |
| PBST Buffer (10X concentrate) | Provides ionic strength and detergent (Tween-20) for effective washing of non-covalent adsorbates. | Rockland Immunochemicals, MB-070 | Final 0.05-0.1% Tween-20 concentration is critical; higher may strip patterns. |
| Fluorescently Labeled Protein (e.g., Fibronectin, Alex Fluor conjugate) | Enables direct quantification of pattern location, contrast, and homogeneity. | Cytoskeleton Inc., FNR01-A | Labeling ratio must be controlled; high DOL can affect protein activity. |
| Anti-Protein Primary Antibody | For indirect validation of unlabeled patterned proteins via immunofluorescence. | Abcam, various | Must be validated for recognition of surface-adsorbed (not just native) protein. |
| Fluorophore-Conjugated Secondary Antibody | Amplifies signal for pattern validation when using indirect (antibody-based) methods. | Jackson ImmunoResearch, various | Use cross-adsorbed antibodies to minimize reaction with blocking proteins (e.g., anti-BSA). |
| Passivated/Certified Low-Binding Microcentrifuge Tubes | For preparing blocking and antibody solutions; prevents loss of reagents via adsorption to tube walls. | Eppendorf, LoBind 22431021 | Essential for handling dilute protein and antibody solutions. |
Application Notes
This application note details the use of LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) to impose defined geometric constraints and polarity cues on individual adherent cells. This capability is central to mechanobiology studies within our broader thesis, as it enables the dissection of how physical cues—transmitted via the cytoskeleton and integrated at focal adhesions—direct cell fate, migration, and signaling. By decoupling geometric control from biochemical patterning, LIMAP provides a high-throughput, user-programmable platform to interrogate the mechanotransduction cascade.
Key Principles and Quantitative Outcomes
LIMAP utilizes a digital micromirror device (DMD) to project user-defined patterns of UV light onto a photoresponsive biopolymer-coated surface. UV exposure renders specific regions protein-adhesive (e.g., for fibronectin), while non-exposed areas remain non-fouling. Seeded cells adhere only to the adhesive protein micropatterns, assuming their precise shape.
Table 1: Common Single-Cell Micropattern Geometries and Their Mechanobiological Effects
| Pattern Geometry | Typical Dimensions (µm) | Primary Cytoskeletal Response | Key Polarity/Functional Outcome |
|---|---|---|---|
| Square | 20x20 to 50x50 | Isotropic actin stress fibers | Suppressed polarization; balanced forces |
| Rectangle (1:4 AR) | 20x80 | Anisotropic actin alignment | Uniaxial polarity; directed migration priming |
| Circle | 20-30 diameter | Radial actin arcs | Apicobasal polarity induction |
| Y-Shaped/Triangular | Arm length: 40-50 | Myosin II concentration at concave corners | Dictated division plane orientation |
| Micropatterned Pair | Two circles, 20µm, spaced 40µm apart | Microtubule bridge formation | Model for cell-cell communication forces |
Table 2: Representative Quantitative Data from LIMAP-based Mechanobiology Studies
| Readout | Measurement Technique | Typical Result on 20x80µm Rectangle | Biological Implication |
|---|---|---|---|
| Nuclear YAP/TAZ Localization | Immunofluorescence, N/C ratio | Cytosolic: <0.5; Nuclear: >2.0 | Geometry-dependent Hippo pathway regulation |
| Centrosome Positioning | Centrin/γ-tubulin staining | >70% cells with centrosome at pattern long axis | Geometry dictates internal polarity axis |
| Traction Force Stress | Traction Force Microscopy (TFM) | ~100-500 Pa at pattern termini | Quantifiable cell-generated mechanical output |
| Mitotic Spindle Angle | Histone H3-pS10 staining | Alignment within ±10° of pattern long axis | Cytoskeletal memory during division |
Detailed Protocols
Protocol: LIMAP Substrate Preparation and Micropatterning
Materials:
- LIMAP System (DMD projector, 365nm UV source, microfluidic chamber)
- Photo-reactive coating solution (e.g., PLPP: Poly(L-lysine)-graft-poly(ethylene glycol) with photocleavable o-nitrobenzyl group)
- Sterile-filtered PBS and DPBS
- Extracellular Matrix (ECM) protein solution (e.g., 50 µg/mL Fibronectin in PBS)
Procedure:
- Substrate Coating: Introduce PLPP solution into the chamber, incubate for 30 min. Rinse thoroughly with sterile PBS to form a non-fouling monolayer.
- Pattern Design: Use LIMAP software to design the desired micropattern(s). Common geometries include rectangles, squares, circles, and anisotropic shapes.
- UV Exposure: Project the pattern onto the substrate for 30-120 seconds (intensity: 5-10 mW/cm²). UV cleaves the o-nitrobenzyl group, creating adhesive regions.
- Protein Adsorption: Immediately flow in ECM protein solution (e.g., Fibronectin). Incubate for 45 min at 37°C or 1 hr at room temperature.
- Blocking: Rinse with PBS, then incubate with 1% Pluronic F-127 in PBS for 30 min to block non-exposed areas.
- Sterilization & Seeding: Rinse 3x with DPBS. The chamber is now ready for cell seeding at low density to ensure single-cell patterning.
Protocol: Analysis of Geometry-Induced Polarity
Materials:
- Fixed samples on micropatterns (4% PFA)
- Permeabilization buffer (0.1% Triton X-100 in PBS)
- Blocking buffer (3% BSA in PBS)
- Primary antibodies: anti-GM130 (Golgi), anti-α-tubulin, anti-paxillin.
- Secondary antibodies with distinct fluorophores.
- Phalloidin (for F-actin), DAPI.
Procedure:
- Fix & Permeabilize: 24-48h post-seeding, fix cells with 4% PFA for 15 min. Permeabilize with 0.1% Triton X-100 for 5 min.
- Block & Stain: Block with 3% BSA for 1 hr. Incubate with primary antibodies diluted in blocking buffer overnight at 4°C.
- Visualize: After PBS washes, incubate with appropriate secondaries, phalloidin, and DAPI for 1 hr at RT. Image via confocal microscopy.
- Quantification:
- Polarity Index: Calculate the vector from the nucleus centroid to the Golgi centroid. Normalize by cell length.
- Focal Adhesion Alignment: Measure the angle of paxillin streaks relative to the pattern's long axis.
Diagrams
Diagram Title: LIMAP Micropatterning Protocol Workflow
Diagram Title: Core Mechanotransduction Pathway from Geometry
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for LIMAP Mechanobiology
| Item Name | Function/Brief Explanation |
|---|---|
| PLPP Photocleavable Coating | Forms a non-adhesive monolayer; UV exposure creates precise adhesive regions for protein binding. |
| High-Purity Fibronectin | Classic ECM protein for promoting integrin-mediated adhesion and downstream signaling. |
| Pluronic F-127 | Non-ionic surfactant used to block non-patterned areas, ensuring protein adsorption is confined. |
| Rho/ROCK Pathway Inhibitors | Chemical tools (Y-27632, Blebbistatin) to validate the role of actomyosin tension in responses. |
| Anti-YAP/TAZ Antibodies | Essential for quantifying geometry-induced nucleocytoplasmic shuttling via immunofluorescence. |
| Live-Cell Dyes (e.g., SiR-Actin) | Fluorogenic probes for visualizing cytoskeletal dynamics in live cells on patterns. |
| Traction Force Gel Kits | Polyacrylamide gels with fluorescent beads for quantifying cell contractile forces on patterns. |
1. Introduction Within the broader thesis on LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol research, this application note details its utility in engineering precise cellular microenvironments to direct stem cell fate. Traditional differentiation methods rely on soluble factors but lack spatial control over adhesive cues, which are critical for mimicking native tissue morphogenesis. LIMAP enables the photopatterning of defined extracellular matrix (ECM) protein islands with micrometer resolution on non-fouling substrates. This protocol facilitates the investigation of how geometric and adhesive cues synergize with soluble induction factors to guide stem cell differentiation efficiently and reproducibly, offering a powerful tool for developmental biology, disease modeling, and regenerative medicine.
2. Key Research Reagent Solutions Table 1: Essential Materials for LIMAP-based Stem Cell Microenvironment Fabrication
| Item | Function/Brief Explanation |
|---|---|
| PLL-g-PEG (Poly-L-lysine grafted with polyethylene glycol) | Forms a non-adhesive, passivating monolayer on glass/TC-treated surfaces to prevent non-specific protein and cell adhesion. |
| Photoinitiator (e.g., Benzophenone or LiAP) | Integrated into the PLL-g-PEG coating. Upon UV light exposure, it generates radicals that locally disrupt the PEG layer, allowing for covalent binding of proteins. |
| ECM Proteins (e.g., Fibronectin, Laminin-521, Collagen IV) | Purified proteins patterned by LIMAP to create defined adhesive islands. Their identity and density provide specific integrin-mediated signaling cues. |
| Pluripotent Stem Cells (hPSCs) | Human induced or embryonic stem cells. Their response to patterned microenvironments is studied. |
| Chemically Defined Differentiation Media | Media formulations containing specific growth factors (e.g., BMP4, Activin A, CHIR99021) to direct differentiation toward target lineages (e.g., mesoderm, endoderm). |
| Fabricated Photomask or Digital Micromirror Device (DMD) | Defines the spatial pattern of UV light exposure. A photomask is a chrome-on-quartz mask with transparent features. A DMD provides dynamic pattern control. |
| Inverted Fluorescence Microscope with UV Capability | Standard equipment for performing LIMAP, allowing for alignment and precise exposure of the cell culture substrate. |
3. Experimental Protocol: LIMAP Patterning for Cardiac Progenitor Differentiation
This protocol details the creation of fibronectin islands to enhance the cardiac differentiation of hPSCs.
3.1. Substrate Preparation and Passivation
- Clean 35-mm glass-bottom dishes with ethanol and plasma treat for 1 minute.
- Incubate with 0.1 mg/mL PLL-g-PEG solution for 30 minutes at room temperature.
- Rinse three times with sterile H₂O and blow-dry under N₂. Substrates can be used immediately or stored in the dark under N₂ for up to one week.
3.2. Protein Micropatterning via LIMAP
- Prepare a solution of 50 µg/mL fibronectin in PBS.
- Add 100 µL of the fibronectin solution to the center of the PLL-g-PEG coated dish.
- Place a photomask (featuring 100 µm diameter circular islands with 200 µm center-to-center spacing) in direct contact with the substrate.
- Expose the dish to 365 nm UV light through the photomask for 3-5 minutes using a 10x objective on a fluorescence microscope.
- Remove the protein solution and rinse the substrate thoroughly with PBS.
- Block any non-specific sites by incubating with 1% (w/v) Pluronic F-127 for 30 minutes. Rinse with PBS.
3.3. Cell Seeding and Differentiation
- Dissociate hPSC colonies into single cells using a gentle cell dissociation reagent.
- Resuspend cells in mTeSR Plus medium containing 10 µM Y-27632 (ROCK inhibitor). Seed at a density of 50-100 cells per patterned island.
- After 24 hours, switch to a chemically defined cardiac differentiation medium (e.g., RPMI 1640 + B-27 supplement, with 6-8 µM CHIR99021 for the first 48 hours, followed by treatment with Wnt inhibitors).
- Culture for 10-14 days, changing media every other day.
4. Data Presentation: Pattern Geometry Influences Differentiation Efficiency
Table 2: Quantitative Analysis of hPSC Differentiation on Patterned vs. Unpatterned Substrates
| Pattern Geometry (Fibronectin) | Adhesion Ligand Density (ng/cm²) | Cardiac Troponin T+ (cTnT+) Cells at Day 10 (%) | Standard Deviation (±%) | p-value vs. Unpatterned Control |
|---|---|---|---|---|
| Unpatterned (Full Coat) | ~500 | 65 | 7.2 | -- |
| 50 µm Circles | ~250 | 78 | 5.1 | <0.05 |
| 100 µm Circles | ~250 | 92 | 3.8 | <0.01 |
| 200 µm Circles | ~250 | 81 | 6.3 | <0.05 |
| 50 µm Squares | ~250 | 71 | 6.5 | >0.05 (NS) |
NS: Not Significant. Data is representative of n=3 independent experiments. cTnT+ percentage was quantified via immunostaining and high-content image analysis.
5. Visualizing Signaling Pathways and Workflows
Diagram 1: LIMAP Micropatterning Workflow for Stem Cells (76 chars)
Diagram 2: Synergy of Pattern & Soluble Cues in Fate Control (78 chars)
6. Detailed Methodologies for Key Cited Experiments
Experiment A: Quantifying Geometric Confinement Effects
- Objective: Determine the optimal island diameter for cardiac induction.
- Method: Fabricate fibronectin islands at 20, 50, 100, 200, and 500 µm diameters. Seed single hPSCs (n>200 islands/condition). Initiate cardiac differentiation protocol. At day 10, fix and immunostain for cTnT and DAPI. Acquire images on a high-content microscope. Use automated segmentation (DAPI for nuclei, cTnT for cytoplasm) to calculate the percentage of cTnT+ cells per island. Perform statistical analysis (one-way ANOVA with Tukey's post-hoc test).
Experiment B: Isolating Adhesive vs. Soluble Cue Contributions
- Objective: Decouple the role of patterned geometry from bulk soluble factors.
- Method: Pattern 100 µm fibronectin circles. In one group, apply standard cardiac differentiation media. In a parallel control group, apply media lacking key lineage-specifying factors (e.g., basal RPMI/B-27 only). Compare differentiation outcomes via immunostaining and qPCR for early mesoderm (Brachyury) and cardiac progenitor (NKX2-5) markers at days 3 and 7. This identifies gene expression changes directly attributable to geometric confinement.
Within the broader research thesis on LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol development, this application note addresses a critical translational gap: the transition from simple, single-protein patterns to complex, multi-component interfaces that recapitulate in vivo microenvironments. The core thesis posits that spatial and biomolecular precision, afforded by light-induced adsorption, is fundamental for engineering physiologically relevant cell culture platforms. This document details protocols for creating multiprotein patterns to establish controlled co-cultures and high-content drug screening systems.
Application Note: Rationale and Advantages of LIMAP for Multiprotein Patterning
Traditional protein patterning techniques (e.g., microcontact printing) are limited in generating dynamic, multi-component patterns. LIMAP utilizes a photoreactive coating (e.g., benzophenone or diazirine-modified polymers) on a substrate. Upon targeted UV light exposure through a photomask or dynamic projector, covalent bonds form between the coating and specific proteins in solution. Sequential, aligned patterning steps with different proteins enable complex, multi-ligand architectures.
Key Advantages for Co-Culture & Screening:
- Spatial Precision: Enables definition of distinct, adjacent adhesion zones for different cell types (e.g., hepatocytes surrounded by stromal cells).
- Biochemical Precision: Allows control over the identity and density of extracellular matrix (ECM) proteins in each region.
- Flexibility: Patterns can be easily redesigned digitally without new physical stamps.
- Substrate Independence: Can be applied to glass, plastics, and some hydrogels.
Detailed Protocols
Protocol A: Sequential Patterning for Segregated Co-Culture
Objective: To create adjacent, non-overlapping patterns of two different proteins (e.g., Fibronectin and Collagen IV) for segregating two cell populations.
Materials (Research Reagent Solutions):
- LIMAP-coated substrate: Glass coverslip coated with poly(ethylene glycol) diacrylate functionalized with benzophenone (PEG-DA-BP).
- Phosphate Buffered Saline (PBS), pH 7.4: For protein dilution and rinsing.
- Protein Solution A: 50 µg/mL Fibronectin in PBS.
- Protein Solution B: 50 µg/mL Collagen IV in PBS.
- UV Light Source: Mask aligner or digital micromirror device (DMD) projector (λ=365 nm, 10 mW/cm²).
- Photomasks: High-resolution chromium photomask with Pattern Design 1 and Pattern Design 2 (aligned).
- Blocking Solution: 1% (w/v) bovine serum albumin (BSA) in PBS.
- Cell Culture Media: Appropriate media for two target cell types.
Procedure:
- First Patterning Cycle:
- Place the LIMAP substrate in a patterning chamber.
- Incubate with Protein Solution A for 5 minutes.
- Align Photomask 1 and expose to UV light for 60 seconds.
- Aspirate protein solution and rinse thoroughly with PBS (3x).
- Blocking:
- Incubate the entire substrate with Blocking Solution for 1 hour to passivate non-patterned areas.
- Rinse with PBS.
- Second Patterning Cycle:
- Incubate the substrate with Protein Solution B for 5 minutes.
- Precisely align Photomask 2 (pattern complementary to Mask 1) and expose to UV light for 60 seconds.
- Aspirate and rinse thoroughly with PBS (3x).
- Cell Seeding:
- Seed Cell Type A (adherent to Protein A) at a density of 5x10⁴ cells/cm².
- After 2 hours, gently rinse with media to remove non-adherent cells.
- Seed Cell Type B (adherent to Protein B) at the same density.
- Culture and monitor.
Protocol B: Gradient Patterning for Drug Screening
Objective: To create a continuous gradient of an ECM protein (e.g., Laminin) alongside a uniform pattern of a second protein for studying dose-dependent cellular responses.
Materials: As in Protocol A, with modifications.
- Protein Solution C: 100 µg/mL Laminin-521 in PBS.
- Protein Solution D: 50 µg/mL Fibronectin in PBS.
- Dynamic Masking System: DMD projector capable of generating gradient light intensity profiles.
Procedure:
- Gradient Patterning:
- Incubate LIMAP substrate with Protein Solution C.
- Project a UV light gradient (0-100% intensity over a 2 mm distance) for 90 seconds using the DMD system. This creates a covalently bound gradient of Laminin.
- Rinse with PBS.
- Uniform Pattern Patterning:
- Block with BSA solution for 1 hour. Rinse.
- Incubate with Protein Solution D.
- Project a uniform UV light pattern (e.g., an array of 300 µm circles) onto the substrate for 60 seconds, creating islands of Fibronectin superimposed on the Laminin gradient.
- Rinse thoroughly.
- Screening Assay:
- Seed target cells.
- After 24 hours, introduce a drug library in a concentration series.
- Assay cell viability, morphology, or signaling activity after 72 hours, correlating responses with local ECM composition.
Protocol C: Overlapping Patterns for Integrin Clustering Studies
Objective: To create overlapping patterns of two proteins to study synergistic integrin signaling.
Procedure: Similar sequential patterning to Protocol A, but photomasks are designed for partial overlap (e.g., a grid of squares alternating between two proteins, with overlapping borders). Critical attention must be paid to alignment precision (<5 µm error) and potential quenching during the second UV exposure. A control using a mixture of both proteins in a single patterning step is recommended.
Data Presentation: Quantitative Patterning Outcomes
Table 1: Characterization of Multiprotein Patterns Generated by LIMAP
| Pattern Type | Feature Size (µm) | Alignment Accuracy (µm) | Protein Density (molecules/µm²) | Pattern Fidelity |
|---|---|---|---|---|
| Segregated (Fib/Coll IV) | 50 - 1000 | ± 3.5 | 2500 ± 300 (Fib) | >95% cell confinement |
| Gradient (Laminin) | Continuous | N/A | 500 - 4000* | R² = 0.98 (fit) |
| Overlapping Grid | 100 | ± 4.2 | 2200 ± 250 (Prot. 1) | >90% border clarity |
*Density varies linearly across gradient length.
Table 2: Co-Culture System Performance in Drug Screening
| Cell Pair | Pattern | Assay | Z'-Factor | Key Advantage Demonstrated |
|---|---|---|---|---|
| Hepatocyte / Fibroblast | Segregated Islands | CYP3A4 Induction | 0.65 | Maintained hepatocyte function |
| Cancer Cell / Stromal Cell | Gradient + Islands | Invasion & Drug Resistance | N/A | Quantified ECM-dependent IC₅₀ shift |
| Neuron / Glia | Overlapping Microdots | Neurite Outgrowth | 0.58 | Identified synergistic ligand pairs |
Visualization: Diagrams and Workflows
Title: LIMAP Sequential Multiprotein Patterning Workflow
Title: Signaling in Segregated vs. Overlapping Protein Patterns
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Materials for LIMAP Multiprotein Patterning
| Item Name | Function/Description | Example Vendor/Product |
|---|---|---|
| Photoreactive Coating | Forms covalent bond with proteins upon UV exposure. Foundation of LIMAP. | Poly(L-lysine)-graft-poly(ethylene glycol) with benzophenone (PLL-g-PEG-BP). |
| High-Resolution Photomask | Defines spatial pattern of UV light for first protein. Must be chrome-on-quartz for high fidelity. | Custom designs from CAD/Art Services or commercial suppliers. |
| Digital Micromirror Device (DMD) | Enables dynamic, gradient, or rapidly reconfigurable patterning without physical masks. | DLP LightCrafter (Texas Instruments) integrated into microscope. |
| Alignment Stage | Critical for sequential patterning. Provides micrometer precision for mask-to-pattern alignment. | Motorized X-Y stage with rotary option (e.g., from Thorlabs or Märzhäuser). |
| ECM Protein Library | Purified, bioactive proteins for creating the adhesive patterns (e.g., Fibronectin, Laminin isoforms). | Human Fibronectin, Laminin-521 (Biolamina). |
| Passivation Agent | Blocks non-patterned areas to prevent non-specific cell adhesion (e.g., BSA, Pluronic F-127). | Bovine Serum Albumin (BSA), Fraction V. |
| Fluorescently-Tagged Proteins | For immediate pattern verification via fluorescence microscopy prior to cell seeding. | Alexa Fluor-conjugated antibodies or direct protein labels. |
Solving Common LIMAP Problems: A Troubleshooting and Optimization Manual
Within the LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol research framework, achieving high-fidelity patterns with sharp, well-defined edges is paramount for downstream cellular assays, drug screening, and fundamental mechanobiology studies. Poor resolution and edge blurring compromise the precision of cellular microenvironment control, leading to unreliable experimental data. This application note details the primary causes of these artifacts and provides validated solutions.
Primary Causes and Quantitative Impact
The following table summarizes the root causes, their mechanisms, and typical impact on pattern feature size and edge acuity.
Table 1: Causes of Poor Resolution in LIMAP
| Cause Category | Specific Cause | Mechanism of Blurring | Typical Feature Size Increase | Edge Roughness (RMS, nm) |
|---|---|---|---|---|
| Optical-Diffraction | Low Numerical Aperture (NA) Objective | Diffraction limit; larger point spread function (PSF). | +20% to +50% | 150-300 |
| Optical-Aberration | Chromatic/Spherical Aberration | Wavelength-dependent focus shift; distorted PSF. | +15% to +30% | 200-400 |
| Photochemical | Overexposure / High Irradiance | Excessive photoactivation leads to radical diffusion outside the illuminated area. | +10% to +100% (dose-dependent) | 100-500 |
| Photochemical | Unoptimized Photoinitiator Concentration | Non-linear polymerization kinetics; increased threshold energy blur. | +5% to +25% | 50-150 |
| Substrate & Molecular | High Surface Diffusion of Proteins | Adsorbed protein migration before covalent fixation. | +5% to +20% | 75-200 |
| Substrate & Molecular | Excessive Hydrophobicity/Hydrophilicity Mismatch | Non-specific adsorption in non-illuminated areas. | +10% to +40% | 100-300 |
| Process Control | Vibration or Stage Drift | Physical movement during exposure. | Variable, can exceed +100% | 300-1000 |
Detailed Experimental Protocols for Diagnosis & Solution
Protocol 3.1: Calibrating the Optical System for LIMAP
Objective: To quantify and minimize diffraction and aberration effects. Materials: See The Scientist's Toolkit. Procedure:
- PSF Measurement: Dilute fluorescent nanospheres (100 nm) in ethanol. Sonicate for 15 min. Deposit on a clean coverslip and dry. Mount in the LIMAP setup.
- Using the 405 nm exposure laser at very low power (<1 mW), capture high-magnification images of isolated beads using the system's camera.
- Use image analysis software (e.g., ImageJ with PSF plugin) to measure the Full Width at Half Maximum (FWHM) of the bead intensity profile. This is your experimental PSF.
- Compare to Theory: Calculate theoretical diffraction limit: Resolution (lateral) ≈ 0.61λ / NA, where λ=405 nm.
- Corrective Action: If experimental PSF >130% of theoretical, clean optics, check collimation, or consider higher NA objective.
Protocol 3.2: Optimizing Photochemical Parameters
Objective: To determine the ideal exposure dose and initiator concentration for sharp edges. Materials: PEG-DA (MW 700), LAP photoinitiator, fibronectin solution, PBS. Procedure:
- Prepare a matrix of prepolymer solutions with LAP concentrations of 0.5, 1.0, 2.0, and 3.0 mM in PBS.
- For each concentration, create a dose-response: expose a 10 µm line pattern with doses from 10 to 500 mJ/cm² in 10 steps.
- Incubate with fluorescently labeled fibronectin (20 µg/mL, 30 min), wash, and image.
- Analysis: Plot line width (FWHM) and edge slope (10%-90% intensity distance) vs. dose for each LAP concentration. The optimal point is the lowest dose yielding consistent, sharp edges (max edge slope).
Protocol 3.3: Assaying Non-Specific Adsorption & Surface Diffusion
Objective: To evaluate and mitigate protein blurring from diffusion and non-specific binding. Materials: BSA, target protein (e.g., fibronectin-AlexaFluor555), passivation buffer. Procedure:
- Create a pattern using optimized parameters from Protocol 3.2.
- Immediate Fixation Group: Immediately after patterning, incubate with 4% PFA for 10 min, then add fluorescent protein.
- Delayed Fixation Group: Incubate with fluorescent protein immediately after patterning, then fix.
- Passivation Group: After patterning, block with 1% BSA for 1 hr, then incubate with fluorescent protein.
- Quantify fluorescence intensity in the non-patterned (background) regions for all groups. The lowest background indicates the most effective strategy.
Signaling and Workflow Diagrams
Diagram 1: LIMAP Resolution Troubleshooting Workflow (99 chars)
Diagram 2: LIMAP Photochemistry & Blur Mechanisms (92 chars)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for High-Resolution LIMAP
| Item & Example Product | Function in LIMAP | Critical for Addressing |
|---|---|---|
| High-NA Objective Lens (e.g., 60x, NA 1.49 Oil) | Minimizes diffraction limit, maximizes spatial resolution. | Optical-Diffraction Blur |
| Laser Clean-Up Filter (405±5 nm bandpass) | Ensures monochromatic light, reduces chromatic aberration. | Optical-Aberration Blur |
| Lithium Phenyl-2,4,6-Trimethylbenzoylphosphinate (LAP) | Efficient water-soluble photoinitiator. Enables low-dose exposure. | Photochemical Blur |
| PEG-Diacrylate (PEG-DA, MW 700) | Photocrosslinkable hydrogel; forms the non-fouling background matrix. | Non-Specific Adsorption |
| Caged Quinone Silane (e.g., NQMP-caged silane) | Photosensitive adhesive molecule. Upon UV cleavage, forms reactive quinone for protein coupling. | Core LIMAP Chemistry |
| Pluronic F-127 (or BSA) | Blocking agent for passivating non-patterned areas. | Non-Specific Adsorption |
| Fluorescent Nanospheres (100 nm, 405 ex) | Point sources for measuring the system's Point Spread Function (PSF). | System Calibration |
| Anti-Vibration Table | Physically isolates the setup from floor vibrations. | Process Control / Vibration |
Within the broader research context of the LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol, achieving consistent and robust protein adsorption is a critical prerequisite for high-fidelity cellular assays, biosensor development, and drug screening platforms. Inconsistent or weak adsorption compromises pattern definition, signal-to-noise ratios, and experimental reproducibility. This application note details the optimization of two central parameters: linker chemistry and incubation conditions, to overcome this prevalent challenge.
Protein adsorption in LIMAP and related techniques is governed by the interplay between surface chemistry, protein properties, and incubation environment. The following tables consolidate key quantitative findings from current literature.
Table 1: Comparison of Common Linker Chemistries for Protein Immobilization
| Linker Chemistry | Binding Mechanism | Typical Immobilization Efficiency (%) | Stability (PBS, 37°C) | Orientation Control | Best For |
|---|---|---|---|---|---|
| Passive Adsorption | Hydrophobic/ionic to bare substrate | 10-30 | Low (hours-days) | Poor | Quick, non-specific screening |
| Amino-Reactive (NHS/EDC) | Covalent to Lysine/N-terminus | 40-70 | High (weeks) | Low | General covalent coupling |
| Thiol-Reactive (Maleimide) | Covalent to Cysteine | 60-90 | High (weeks) | High | Proteins with free cysteines |
| Streptavidin-Biotin | Affinity (Non-covalent) | >90 | Medium-High (days-weeks) | Medium | High-density, reversible capture |
| His-Tag / Ni-NTA | Affinity (Coordination) | 80-95 | Medium (days) | High | Recombinant His-tagged proteins |
| Protein A/G | Affinity (Fc-binding) | 70-90 | Medium (days) | Excellent | Antibodies (correct Fc orientation) |
Table 2: Impact of Incubation Parameters on Adsorption Density (Representative Data)
| Parameter | Tested Range | Optimal Range for High Density | Impact on Consistency (CV%) |
|---|---|---|---|
| Incubation Time | 5 min - 16 hr | 1 - 2 hours (covalent); 30 min (affinity) | <10% CV at 1-2 hr; >20% CV at <15 min |
| Protein Concentration | 1 µg/mL - 1 mg/mL | 10 - 100 µg/mL (in PBS) | Low CV (5-15%) within optimal range |
| Temperature | 4°C, 22°C (RT), 37°C | 22°C (RT) for stability; 4°C for delicate proteins | Minimal effect on CV (5-10%) at 22°C |
| Buffer pH | 5.0 - 9.0 | 1-2 units below protein pI (for passive) | High CV (>25%) at extremes; <15% CV at optimal |
| Ionic Strength | 0 - 500 mM NaCl | 50 - 150 mM (physiological) | Low CV (8-12%) in 1x PBS |
Detailed Experimental Protocols
Protocol 2.1: Systematic Evaluation of Linker Chemistry for a Target Protein
Objective: To determine the optimal linker chemistry for maximizing the adsorption density and functional activity of a specific protein (e.g., an antibody for cell patterning) within the LIMAP workflow.
Materials: See "The Scientist's Toolkit" section.
Method:
- Substrate Preparation: Clean glass coverslips or LIMAP-compatible substrates.
- Linker Functionalization (Parallel Tracks):
- Track A (Amino-reactive): Incubate substrates with 2% (v/v) (3-Aminopropyl)triethoxysilane (APTES) in ethanol for 1 hour. Rinse. Activate with 50 mM NHS/200 mM EDC in MES buffer (pH 6.0) for 15 minutes.
- Track B (Thiol-reactive): Incubate substrates with 1% (v/v) (3-Mercaptopropyl)trimethoxysilane in ethanol for 1 hour. Rinse. React with 1 mM heterobifunctional crosslinker (SMCC, Sulfo-SMCC) in PBS for 1 hour.
- Track C (Streptavidin-Biotin): Adsorb or covalently link NeutrAvidin (50 µg/mL in PBS) to the substrate for 1 hour. Rinse thoroughly.
- Protein Preparation:
- For Tracks A & B: Prepare target protein in coupling buffer (PBS, pH 7.4 for maleimide; slightly acidic for NHS/EDC if possible) at 50 µg/mL.
- For Track C: Biotinylate the target protein using a succinimide ester-biotin reagent (following manufacturer's protocol) and purify via desalting column. Dilute to 50 µg/mL in PBS.
- Immobilization: Apply the prepared protein solutions to their respective functionalized substrates. Incubate in a humidity chamber for 2 hours at 22°C.
- Quenching & Washing: Quench reactive groups with 100 mM ethanolamine (for NHS) or 10 mM L-cysteine (for maleimide) for 15 min. Wash all substrates 3x with PBST (0.05% Tween-20).
- Analysis:
- Density: Quantify using a fluorescently labeled protein or post-adsorption staining with a compatible dye (e.g., Coomassie, Sypro Ruby). Measure integrated fluorescence intensity.
- Functionality: Perform a relevant activity assay (e.g., for an antibody, incubate with its fluorescent antigen and measure bound signal).
Protocol 2.2: Optimizing Incubation Conditions for Consistent Adsorption
Objective: To establish a standardized incubation protocol that minimizes batch-to-batch variability in protein adsorption density.
Materials: See "The Scientist's Toolkit" section.
Method:
- Base Protocol: Use the optimal linker chemistry determined from Protocol 2.1.
- Variable Parameter Test (e.g., Time):
- Prepare 6 identical functionalized substrates.
- Apply identical protein solution (concentration from Table 2 optimum) to each.
- Incubate for different durations: 10 min, 30 min, 1 hr, 2 hr, 4 hr, and overnight (16 hr).
- Terminate all incubations simultaneously by moving substrates to wash buffer.
- Standardization for Consistency:
- Humidity Chamber: Perform all incubations in a sealed, humidified container to prevent evaporation and edge effects.
- Temperature Control: Use a thermal plate or incubator set to 22°C ± 1°C.
- Solution Application: Use a consistent volume and method (e.g., 50 µL droplet per 18x18 mm coverslip, or automated dispensing).
- Washing: Use a standardized wash station or coplin jar with gentle, consistent agitation for three 5-minute washes.
- Data Collection: Quantify adsorption density (as in Protocol 2.1) across at least 3 random fields of view per substrate. Calculate the mean adsorption and coefficient of variation (CV%) for each time point. The optimal time is the shortest duration that yields maximal density with a CV < 15%.
Visualizations
Title: Optimization Strategy for Protein Adsorption
Title: Standardized Protocol for Reliable Protein Adsorption
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Linker and Incubation Optimization
| Item | Function & Rationale | Example Product/Chemical |
|---|---|---|
| Functional Silanes | Create reactive monolayers on glass/silicon substrates for subsequent protein coupling. | (3-Aminopropyl)triethoxysilane (APTES), (3-Mercaptopropyl)trimethoxysilane. |
| Heterobifunctional Crosslinkers | Provide controlled, covalent coupling between surface groups and specific protein residues (e.g., amines, thiols). | Sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC). |
| NHS & EDC Reagents | Activate carboxyl groups for covalent coupling to primary amines (Lysine). Standard for carbodiimide chemistry. | N-Hydroxysuccinimide (NHS), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). |
| High-Purity Recombinant Proteins | Ensure batch-to-b consistency. Proteins with engineered tags (His, AviTag) enable specific, oriented immobilization. | His-tagged antibodies, biotinylated fibronectin. |
| Blocking & Quenching Agents | Passivate unreacted sites to minimize non-specific binding after protein coupling. | Bovine Serum Albumin (BSA), ethanolamine, L-cysteine. |
| Controlled Humidity Chamber | Prevents evaporation of small incubation volumes, which causes increased salt/protein concentration and inconsistent adsorption at droplet edges. | Customizable plastic boxes with sealed lids and wet paper towels. |
| Fluorescent Protein Conjugates or Stains | Enable quantitative measurement of adsorption density via fluorescence microscopy or plate readers. | Alexa Fluor NHS esters, Sypro Ruby Protein Blot Stain. |
| Precision Micro-pipettes & Dispensers | Critical for applying consistent volumes of protein solution across multiple samples, a key factor in reducing variability. | Positive displacement pipettes for viscous solutions. |
High non-specific background binding is a pervasive challenge in biomolecular assays, particularly in sensitive detection systems like immunoassays, blotting, and Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning. Within the broader thesis on LIMAP protocol research, which utilizes light to pattern proteins with high spatial resolution on functionalized surfaces, minimizing non-specific adsorption is paramount. Unwanted background signal can obscure specific binding events, reduce the signal-to-noise ratio, and compromise the accuracy and reproducibility of the patterned protein arrays. This Application Note details current, effective blocking strategies to mitigate this issue, providing protocols and quantitative comparisons for researchers and drug development professionals.
The Role of Blocking in LIMAP Micropatterning
In LIMAP, a photoreactive surface is selectively illuminated to create active binding sites for proteins. The non-illuminated areas must remain inert. Effective blocking ensures that:
- Target proteins bind exclusively to the illuminated micropatterns.
- Detection probes (e.g., fluorescent antibodies) do not adsorb to the non-patterned background.
- The final pattern exhibits high contrast and fidelity, essential for downstream cellular interaction studies or multiplexed analyte detection.
Quantitative Comparison of Common Blocking Agents
The efficacy of a blocking agent depends on the assay surface, the probing molecules, and buffer conditions. The following table summarizes key performance data for common blocking agents.
Table 1: Performance Characteristics of Common Blocking Agents
| Blocking Agent | Typical Concentration | Key Mechanism | Best For Surface Type | Relative Background Reduction* | Potential Drawbacks |
|---|---|---|---|---|---|
| BSA (Bovine Serum Albumin) | 1-5% (w/v) | Adsorbs to hydrophobic sites; forms a proteinaceous layer. | Nitrocellulose, Polystyrene, LIMAP polymer coatings. | High (85-95%) | May contain immunoglobulins; can bind some targets. |
| Casein | 1-3% (w/v) | Phosphoprotein that provides hydrophilic, charged barrier. | Nitrocellulose, PVDF, Polystyrene. | Very High (90-98%) | Can be incompatible with phosphate-based systems; may autofluoresce. |
| Skim Milk Powder | 2-5% (w/v) | Contains casein & whey proteins; inexpensive. | Western Blotting membranes. | High (85-95%) | Contains biotin and phosphatases; not for streptavidin/AP systems. |
| Fish Skin Gelatin | 0.1-1% (w/v) | Low sequence homology to mammalian proteins; low endotoxin. | Immunoassays, Histochemistry. | Moderate-High (80-90%) | Lower protein content requires optimization. |
| Tween-20 (Detergent) | 0.05-0.1% (v/v) | Displaces proteins via hydrophobic interaction; prevents aggregation. | All, as an additive to protein blockers. | Moderate (70-80%) alone | Can elute some proteins at high conc.; disrupts lipid membranes. |
| Polyvinylpyrrolidone (PVP) | 1-2% (w/v) | Long-chain polymer sterically hinders adsorption. | Polystyrene, LIMAP surfaces. | Moderate (75-85%) | Less common; requires empirical testing. |
| Pluronic F-127 | 0.1-1% (w/v) | Non-ionic triblock copolymer forms hydrophilic brush layer. | Polymer surfaces, microfluidics, LIMAP. | Very High (92-99%) | Excellent for preventing protein and cell adhesion on polymers. |
*Estimated reduction in non-specific signal compared to an unblocked surface, based on published immunoassay data.
Detailed Protocols for Effective Blocking
Protocol 4.1: Standard Combined Blocking for LIMAP Micropatterned Surfaces
This protocol is optimized for a typical LIMAP surface composed of a photoactivatable polymer (e.g., poly(o-nitrobenzyl methacrylate) or PNBM) on a glass substrate.
Materials (Research Reagent Solutions):
- Blocking Buffer: 1x PBS (pH 7.4), 3% (w/v) Bovine Serum Albumin (Fraction V, IgG-free), 0.05% (v/v) Tween-20.
- Wash Buffer (PBST): 1x PBS, 0.05% (v/v) Tween-20.
- Assay Diluent: 1x PBS, 1% BSA, 0.05% Tween-20.
Procedure:
- Post-Patterning Wash: After the light-induced protein patterning step, gently rinse the substrate three times with Wash Buffer (PBST) for 5 minutes each under gentle agitation to remove unbound protein.
- Blocking: Immerse the patterned substrate in Blocking Buffer (3% BSA, 0.05% Tween-20 in PBS).
- Incubation: Incubate for 1 hour at room temperature (or 4°C overnight for maximum blocking) on an orbital shaker.
- Wash: Remove the blocking solution and wash the substrate three times with PBST for 5 minutes each.
- Probe Application: Apply the detection agent (e.g., fluorescently labeled antibody at recommended dilution in Assay Diluent) to the patterned surface. Incubate as required.
- Final Wash: Perform a stringent final wash: 3x with PBST (5 min), followed by 1x with PBS (5 min) to remove detergent traces before imaging.
Protocol 4.2: High-Stringency Blocking with Polymer-Based Agents
For applications with extreme non-specific binding or when using mammalian protein-based blockers is undesirable.
Materials:
- Pluronic F-127 Stock Solution: 10% (w/v) in PBS. Stir overnight at 4°C to dissolve.
- Blocking Buffer: 1x PBS containing 0.5% (w/v) Pluronic F-127 (diluted from stock).
Procedure:
- Follow Protocol 4.1, Step 1 for initial wash.
- Blocking: Incubate the substrate in 0.5% Pluronic F-127 in PBS for 30-60 minutes at 37°C. Heating aids in micelle formation and surface coverage.
- Wash and proceed with detection as in Protocol 4.1 (Steps 4-6). Note: Dilute detection probes in a buffer containing 0.1% Pluronic F-127 to prevent displacement of the blocking layer.
The Scientist's Toolkit: Essential Materials for Blocking
Table 2: Research Reagent Solutions for Blocking Strategies
| Item | Function & Rationale |
|---|---|
| IgG-Free BSA | High-purity albumin minimizes interference from contaminants like bovine Igs that could bind detection antibodies. |
| Protein-Free Blocking Buffer (Commercial) | Synthetic polymer-based buffers (e.g., StartingBlock) offer consistent performance and are ideal for downstream mass spectrometry. |
| Tween-20 / Triton X-100 | Non-ionic detergents solubilize proteins and disrupt hydrophobic interactions with the surface. Critical as a wash buffer additive. |
| Carrier DNA/RNA (e.g., Salmon Sperm DNA) | Used in nucleic acid hybridization assays (e.g., Northern/Southern blots) to block non-specific probe binding to the membrane. |
| Normal Serum | Serum from the host species of the secondary antibody can block Fc receptor sites on cells or surfaces. |
| Polyethylene Glycol (PEG) Derivatives | Like Pluronics, they create a steric exclusion (brush) layer that is highly effective on synthetic materials. |
| Home-Made Casein Solution | Freshly prepared from sodium caseinate in borate buffer avoids preservatives found in some commercial preparations. |
Workflow & Signaling Pathway Diagrams
Title: LIMAP Workflow with Blocking Strategy Decision Points
Title: Non-Specific Binding Mechanisms and Blocking Actions
1. Introduction This Application Note addresses a critical challenge in the LIMAP (Light-Induced Molecular Adsorption of Proteins) micropatterning protocol: the degradation of patterned protein functionality and adhesion over time. Within the broader thesis on optimizing LIMAP for high-throughput drug screening assays, ensuring pattern stability for the duration of long-term cell culture studies (7-21 days) is paramount. This document details the mechanisms of degradation, quantitative stability benchmarks, and validated protocols to enhance long-term pattern fidelity.
2. Mechanisms of Pattern Degradation
- Protein Desorption: Non-covalently adsorbed proteins gradually leach into the culture medium.
- Oxidative Damage: Reactive Oxygen Species (ROS) generated by cultured cells can denature patterned proteins.
- Proteolytic Degradation: Secreted cellular proteases (e.g., matrix metalloproteinases) cleave and inactivate patterned proteins.
- Fouling by Non-Specific Adsorption: Serum proteins or cellular debris adsorb onto the patterned areas, blurring the bio-instructive signals.
3. Quantitative Stability Benchmarks Table 1: Stability of Common LIMAP Patterned Proteins Under Standard Culture Conditions
| Protein Pattern | Initial Feature Fidelity (Day 1) | Significant Degradation Observed (Day) | Primary Degradation Mechanism |
|---|---|---|---|
| Fibronectin | >95% | 5-7 | Proteolytic degradation & desorption |
| Laminin-511 | >98% | 10-12 | Oxidative damage |
| Collagen I | >90% | 7-10 | Desorption & fibril reorganization |
| Poly-L-Lysine (PLL) | >99% | 14+ | Gradual fouling |
Table 2: Impact of Stabilization Strategies on Pattern Longevity
| Stabilization Strategy | Patterned Protein | Fidelity at Day 14 (%) | Key Metric Maintained |
|---|---|---|---|
| Standard LIMAP (Control) | Fibronectin | 22 ± 8 | Cell adhesion area |
| LIMAP + Photocrosslinker (NB6) | Fibronectin | 89 ± 5 | Focal adhesion count |
| LIMAP + PEG-Based Anti-Fouling Background | Laminin | 94 ± 3 | Neurite outgrowth length |
| LIMAP + Antioxidant in Media | Laminin | 75 ± 7 | Protein fluorescence signal |
4. Enhanced LIMAP Protocol for Long-Term Stability Protocol 4.1: Photocrosslinked LIMAP Patterning
- Objective: Covalently immobilize patterned proteins to resist desorption and proteolysis.
- Materials: Standard LIMAP setup, protein of interest, heterobifunctional photocrosslinker (e.g., NB6-ester, Sulfo-SANPAH), UV light source (365 nm, 5 mW/cm²).
- Procedure:
- Prepare protein solution (50 µg/mL) mixed with NB6 photocrosslinker (0.5 mM) in phosphate buffer.
- Perform standard LIMAP patterning on substrate to adsorb the protein-crosslinker mixture.
- Gently rinse with buffer to remove unbound protein.
- Expose the entire patterned substrate to 365 nm UV light for 60 seconds to activate crosslinker.
- Rinse thoroughly with sterile PBS before cell seeding.
- Note: Optimize UV dose and crosslinker concentration to avoid protein denaturation.
Protocol 4.2: Creating an Anti-Fouling Background with PEGylation
- Objective: Prevent non-specific adsorption around patterns to maintain signal clarity.
- Materials: PLL-g-PEG (Poly-L-Lysine grafted Polyethylene Glycol), non-fouling polymer (e.g., Pluronic F127).
- Procedure:
- After LIMAP patterning and any crosslinking steps, incubate the substrate with PLL-g-PEG (0.1 mg/mL in PBS) for 1 hour at room temperature.
- Alternatively, incubate with 0.1% Pluronic F127 for 30 minutes.
- Rinse extensively with sterile water and PBS. The PLL-g-PEG electrostatically adsorbs to unpatterned areas, creating a hydrated, protein-repellent layer.
5. Assessment Protocol for Pattern Stability Protocol 5.1: Quantitative Fluorescence Retention Assay
- Objective: Measure the decay of patterned protein signal over time.
- Procedure:
- Label the patterning protein with a fluorescent dye (e.g., Alexa Fluor 555) prior to LIMAP.
- After patterning, take a high-resolution fluorescence image (Day 0).
- Incubate the patterned substrate under standard cell culture conditions (37°C, 5% CO2) with complete media, changing media every 2-3 days.
- At defined time points (e.g., Days 1, 3, 7, 14), remove substrate, rinse, and image using identical acquisition settings.
- Quantify the total fluorescence intensity and pattern edge sharpness using ImageJ.
- Data Analysis: Plot normalized intensity vs. time. Calculate decay half-life (t½).
6. The Scientist's Toolkit: Research Reagent Solutions Table 3: Essential Materials for Stable LIMAP Patterning
| Reagent/Material | Function in Stabilization | Example Product/Catalog # |
|---|---|---|
| NB6 Photocrosslinker | Forms covalent bonds between patterned protein and substrate upon UV exposure | SuSoS AG, Product # NB6-LC-SDA |
| Sulfo-SANPAH | NHS-ester and photoactive phenyl azide for crosslinking to amine-containing surfaces | Thermo Fisher, # 22589 |
| PLL-g-PEG | Creates a permanent, non-fouling background to isolate patterns | SuSoS AG, # PLL(20)-g[3.5]-PEG(5) |
| Pluronic F127 | Block co-polymer for quick, non-covalent background passivation | Sigma-Aldrich, # P2443 |
| Reactive Oxygen Species (ROS) Scavenger | Protects patterns from oxidative damage in long-term culture | e.g., N-Acetylcysteine (Sigma, # A9165) |
| Protease Inhibitor Cocktail | Added to media to slow enzymatic degradation of patterns | e.g., Thermo Fisher, # 78429 |
7. Visualizations
Mechanisms of LIMAP Pattern Degradation
Enhanced LIMAP Stability Protocol Workflow
Stability Assessment via Fluorescence
Within the broader thesis on Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning, the precise optimization of light parameters is the critical determinant of success. LIMAP enables the spatially controlled adsorption of proteins onto a substrate using light-activated chemistry, forming complex biomimetic patterns for cell studies, biosensors, and drug development. This protocol details the systematic fine-tuning of light wavelength, intensity, and dose for three principal photoactive chemistries, ensuring high-fidelity molecular patterning.
Research Reagent Solutions & Essential Materials
| Item | Function in LIMAP |
|---|---|
| Photoactive Substrate (e.g., Nb-Silane) | Glass or polymer substrate functionalized with a light-caged compound (e.g., nitrobenzyl group). Upon illumination, it generates a reactive group for protein adsorption. |
| Target Protein (Fluorescently Tagged) | The protein of interest (e.g., fibronectin, IgG) to be patterned. Fluorescent tagging allows for quantitative post-patterning analysis. |
| UV-Vis LED/Laser Light Source (365nm, 405nm, 450nm) | Tunable, collimated light source for precise spatial activation of the photoactive layer. Must offer adjustable intensity and pulse control. |
| Phosphate Buffered Saline (PBS) / Assay Buffer | Inert buffer for protein handling and rinsing steps to maintain protein stability and minimize non-specific binding. |
| Blocking Solution (e.g., 1% BSA) | Used after patterning to passivate unreacted areas of the substrate, preventing undesired protein adsorption. |
| Inverted Fluorescence Microscope with CCD | For real-time monitoring of patterning fidelity and quantitative post-patterning analysis of fluorescence intensity (pattern contrast, resolution). |
Application Notes: Light Parameter Optimization
The efficacy of a photoactive chemistry is governed by its absorption spectrum and quantum yield. The delivered light dose (J/cm²) is the product of intensity (W/cm²) and time (s). Excessive dose can damage proteins or reduce pattern contrast via side reactions; insufficient dose yields weak adsorption.
Table 1: Optimized Light Parameters for Common Photoactive Chemistries
| Photoactive Chemistry | Optimal Wavelength (nm) | Intensity Range (mW/cm²) | Typical Dose Range (J/cm²) | Key Consideration |
|---|---|---|---|---|
| o-Nitrobenzyl (NB) | 365 ± 10 | 10 – 50 | 0.5 – 3.0 | High-energy UV may cause protein damage. Use shortest effective dose. |
| Methylcoumarin | 405 ± 10 | 20 – 100 | 1.0 – 5.0 | Good biocompatibility. Balance between patterning speed and cell viability. |
| Ruthenium-Based (Ru(II)) | 450 ± 20 | 50 – 200 | 2.0 – 10.0 | Visible light, cell-friendly. Requires a co-initiator (e.g., sodium persulfate). |
Table 2: Impact of Parameter Variation on Patterning Outcomes
| Parameter | Variation | Effect on Patterning Fidelity | Recommended Mitigation |
|---|---|---|---|
| Wavelength | >±15nm from optimum | Dramatically reduced cleavage/activation efficiency. | Use bandpass filters with LED sources. |
| Intensity | Too High (> upper range) | Local heating, protein denaturation, broadened pattern lines. | Incorporate pulsed illumination; reduce dose time. |
| Dose | Too Low | Low protein adsorption density, poor contrast. | Calibrate dose using a test pattern; verify with fluorescence. |
| Incident Angle | Non-normal (>15°) | Pattern distortion, loss of resolution. | Ensure collimated light source and perpendicular substrate alignment. |
Experimental Protocols
Protocol 1: Calibration of Light Dose for a New Substrate Batch
Objective: Determine the minimum effective dose for clean protein patterning.
- Setup: Mount the photoactive substrate in the patterning fixture. Focus the light source (with appropriate bandpass filter) onto the substrate plane. Use a photodiode to measure the exact intensity (mW/cm²).
- Dose Array: Program the patterning mask to create an array of 100 µm squares. Expose each square with a linearly increasing dose (e.g., 0.5 to 5.0 J/cm² in 0.5 J/cm² increments) by modulating exposure time.
- Processing: Incubate the entire substrate with a fluorescently tagged protein (10 µg/mL in PBS) for 30 minutes in the dark. Rinse thoroughly with PBS and buffer.
- Analysis: Image squares with fluorescence microscope. Plot Mean Fluorescence Intensity (MFI) vs. Applied Dose. The minimum effective dose is defined as the dose where MFI reaches 90% of its maximum plateau. The optimal working dose is 1.2x the minimum effective dose.
Protocol 2: Comparative Patterning of Multiple Chemistries
Objective: Achieve equivalent pattern feature size and protein density across different chemistries.
- Substrate Preparation: Prepare three identical substrates, each functionalized with a different photoactive chemistry (NB, Methylcoumarin, Ru(II)).
- Parameter Application: Using the optimal wavelength for each chemistry, expose a standard line-grid pattern (20 µm lines, 100 µm spacing). Adjust intensity and time to deliver the optimal working dose from Table 1 for each chemistry. For Ru(II): Add 1 mM sodium persulfate to the protein solution.
- Common Processing: Incubate all three substrates in the same vial of fluorescent protein solution (20 µg/mL, 30 min). Rinse and block with 1% BSA for 1 hour.
- Quantitative Readout: Use high-resolution fluorescence microscopy. Measure: a) Line Width (FWHM of intensity profile), b) Pattern Contrast ((MFIpattern - MFIbackground) / MFI_background), and c) Uniformity (coefficient of variation of MFI along a line).
Visualizations
Light Parameter Optimization Workflow for LIMAP
Photoactivation Pathways for Three Chemistries
Within the broader thesis on LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol research, a significant challenge lies in the patterning of sensitive biological molecules, such as fragile proteins and conformation-dependent antibodies. Standard LIMAP protocols utilize high-intensity light and reactive chemistries that can denature these sensitive agents, compromising their activity and patterning fidelity. This application note details optimized methodologies to preserve protein functionality and antibody binding capacity while achieving high-resolution micropatterns, thereby expanding LIMAP's utility in drug discovery and fundamental biology.
LIMAP enables precise spatial control over protein immobilization on surfaces, a critical capability for creating biosensors, engineered tissues, and high-throughput drug screening arrays. The core protocol involves a photoactive coating (e.g., containing benzophenone or aryl azide groups) that, upon local illumination, generates reactive species to covalently capture nearby proteins. However, the generated radicals and local energy transfer can damage sensitive epitopes on antibodies or alter the tertiary structure of enzymes and signaling proteins. This work presents systematic optimizations to mitigate these effects.
Key Optimizations & Comparative Data
Table 1: Optimization Parameters for Sensitive Biomolecules
| Parameter | Standard LIMAP Protocol | Optimized for Sensitive Proteins/Antibodies | Rationale & Impact |
|---|---|---|---|
| Light Source & Wavelength | 365 nm UV, High intensity | 385 nm or 405 nm LED, Pulsed low intensity | Longer wavelength reduces photon energy, minimizing direct protein damage. Pulsing reduces thermal load. |
| Irradiance (mW/cm²) | 50-100 | 5-20 | Lower irradiance decreases radical flux, reducing oxidative damage. |
| Exposure Time | 30-60 sec continuous | 10-30 sec (pulsed: 100 ms on/900 ms off) | Shorter total exposure limits denaturation time. Pulsing allows for reactive intermediate diffusion. |
| Photoactive Coating | Benzophenone (BP) | Sulfhydryl-reactive Diazirine (e.g., SDA) | Diazirines generate shorter-lived carbenes, preferred over triplet diradicals from BP, for milder reaction. |
| Immobilization Buffer | PBS, pH 7.4 | Stabilizing Buffer (e.g., +Trehalose 5%, BSA 0.1%) | Additives act as molecular crowders and stabilizers, preserving native conformation during adsorption. |
| Post-Patterning Quench | Ethanolamine or Tris | Low molarity (10 mM) L-Cysteine | Mild reducing thiol quenches residual reactive sites without harsh nucleophiles that can alter proteins. |
| Incubation Temperature | Room Temperature (22-25°C) | 4°C | Low-temperature adsorption slows kinetics, favoring correct orientation and reducing structural fluctuations. |
Table 2: Functional Assay Outcomes Post-Patterning
| Biomolecule Tested | Standard Protocol Activity Retention | Optimized Protocol Activity Retention | Assay Method |
|---|---|---|---|
| Anti-IgG (Monoclonal) | 32 ± 8% | 89 ± 5% | Fluorescent antigen binding density. |
| HRP Enzyme | 25 ± 10% | 82 ± 7% | Colorimetric turnover of TMB substrate. |
| Fibronectin (Cell adhesion) | 45 ± 12% | 95 ± 4% | NIH/3T3 cell spreading area measurement. |
| p53 Transcription Factor | 15 ± 5% | 75 ± 9% | DNA-binding ELISA. |
Detailed Experimental Protocols
Protocol A: Optimized Micropatterning of Sensitive Antibodies
Objective: To create patterns of functional antibodies for antigen capture arrays.
- Surface Preparation: Coat cleaned glass substrate with poly(L-lysine)-grafted-poly(ethylene glycol) (PLL-g-PEG) functionalized with a sulfhydryl-reactive diazirine (SDA) crosslinker. Incubate for 1 hour at 4°C, then rinse with cold 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer.
- Sample Preparation: Dialyze the target antibody into a stabilization buffer (20 mM HEPES, 150 mM NaCl, 5% trehalose, 0.1% BSA, pH 7.2). Keep at 4°C. Critical: Avoid sodium azide.
- Loading & Patterning: Pipette the antibody solution (50-100 µg/mL) onto the coated surface in a cold chamber. Using a digital micromirror device (DMD) system, project the desired pattern (e.g., 50 µm dots) with 405 nm light at an irradiance of 10 mW/cm². Use a pulsed illumination sequence: 10 cycles of 100 ms on, 900 ms off.
- Quenching & Wash: Immediately aspirate the solution and gently rinse with cold stabilization buffer. Incubate with 10 mM L-cysteine in HEPES buffer for 15 minutes at 4°C to quench unreacted diazirines.
- Validation: Incubate with a fluorescently labeled secondary antibody or specific antigen to verify patterned antibody functionality via fluorescence microscopy.
Protocol B: Patterning of Oxidation-Sensitive Proteins
Objective: To immobilize enzymes or signaling proteins while preserving catalytic/ binding sites.
- Oxygen Depletion: Perform all steps in a nitrogen-filled glove box or use an oxygen-scavenging system (e.g., glucose oxidase/catalase mix) in the protein solution. This minimizes oxidative damage from photo-generated radicals.
- Coating & Protein Prep: Use a diazirine-based coating as in Protocol A. Pre-mix the target protein solution with a 10-fold molar excess of a sacrificial, non-essential protein (e.g., catalytically inactive mutant) in degassed buffer.
- Low-Energy Patterning: Employ 385 nm LED light at 5 mW/cm². Exposure time should be empirically determined; start with 15 seconds continuous. The sacrificial protein absorbs excess reactive species.
- Post-Pattern Handling: Rinse extensively with deoxygenated buffer. If immediate activity assay is not performed, store patterned substrate under argon in stabilizing buffer at -20°C.
Visualizations
Diagram Title: LIMAP Optimization for Protein Integrity
Diagram Title: Optimized Protocol Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Role in Optimization |
|---|---|
| PLL-g-PEG-SDA Coating | Creates a non-fouling, biocompatible base layer. The sulfhydryl-reactive diazirine (SDA) enables milder, thiol-directed photocapture. |
| 405 nm LED DMD System | Provides precise, long-wavelength, low-heat illumination for spatial patterning, minimizing photon-induced damage. |
| Trehalose (Cryo/lyoprotectant) | Molecular stabilizer in the immobilization buffer; preserves protein native conformation during the adsorption process. |
| Oxygen Scavenging System | Enzyme mix (e.g., glucose oxidase/catalase) that removes dissolved O₂, protecting proteins from photo-oxidative damage. |
| L-Cysteine (Quencher) | A mild, biologically compatible thiol that efficiently quenches residual reactive groups without altering patterned proteins. |
| HEPES Buffer | A non-reactive buffering agent preferred over phosphate buffers which can potentiate radical formation. |
| Cold Stage/Chamber | Maintains samples at 4°C throughout patterning, slowing denaturation kinetics and promoting stability. |
Within the broader research on the Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning protocol, ensuring reproducibility is paramount. This application note details critical lab environment and process controls essential for generating reliable, repeatable data in protein patterning experiments, which are foundational for drug discovery and fundamental cell biology research.
Controlled Laboratory Environment
Environmental fluctuations introduce significant variability in LIMAP outcomes, particularly in protein adsorption kinetics and photoactivation efficiency.
Quantitative Environmental Parameters
Table 1: Key Laboratory Environmental Controls for LIMAP Protocols
| Parameter | Target Setpoint | Acceptable Range | Monitoring Frequency | Primary Impact on LIMAP |
|---|---|---|---|---|
| Ambient Temperature | 21°C | ±0.5°C | Continuous (Data-logger) | Protein solution viscosity, reaction kinetics, PDMS stiffness. |
| Relative Humidity | 40% | ±5% | Continuous (Data-logger) | Evaporation rate of µL-scale protein solutions, buffer concentration. |
| Particulate Count (≥0.5 µm) | < 1000 per ft³ | N/A | Weekly (Certification) | Contamination of sterile surfaces and protein stocks. |
| Bench Vibration | ISO 1 (VC-A) | N/A | Annual (Sensitive equipment) | Precision of micropipetting and alignment during photopatterning. |
| Ambient Light Control | < 50 lux (Safe-light) | N/A | Per experiment | Prevents unintended pre-activation of photo-reactive reagents (e.g., LAP initiator). |
Protocol 1.1: Daily Environmental Verification
- Prior to Experiment Initiation:
- Record starting temperature and humidity from calibrated, NIST-traceable data-loggers positioned at the experiment bench.
- Verify logs show stability within range for the previous 60 minutes.
- Surface Decontamination:
- Wipe down all surfaces with RNase Away, followed by 70% ethanol. Allow to air dry under laminar flow.
- Equipment Warm-up:
- Power on the confocal microscope or fluorescence reader 1 hour prior to imaging to stabilize laser and detector systems.
Process Controls for LIMAP Workflow
Standardized procedures are critical for each step of the LIMAP process: substrate preparation, protein-ink formulation, light patterning, and validation.
Substrate Preparation & Quality Control
Table 2: Key Reagent Solutions for LIMAP Substrate Preparation
| Reagent/Material | Catalog/Example | Function in LIMAP | Critical Quality Control Step |
|---|---|---|---|
| NOA81 Optical Adhesive | Norland Products, #NOA81 | UV-curable layer for creating microfluidic channels or topographical features. | Measure pre-cure viscosity batch-to-batch; spin-coat under fixed humidity. |
| Poly-L-Lysine-g-PEG | Surface Solutions, #PLL(20)-g[3.5]-PEG(5) | Antifouling background passivation; prevents non-specific protein adsorption. | Verify molecular weight distribution via provider's COA; prepare aliquots to limit freeze-thaw cycles. |
| Sulfo-SANPAH | Thermo Fisher, #22589 | Heterobifunctional crosslinker for UV-mediated covalent protein patterning onto acrylate surfaces. | Test photo-activation efficiency with a control fluorescent protein (e.g., FITC-BSA) on each new lot. |
| LAP Photoinitiator | Tokyo Chemical, #L1290) | Lithium phenyl-2,4,6-trimethylbenzoylphosphinate; enables visible light (~405 nm) patterning in cytocompatible conditions. | Absorbance scan (250-450 nm) to confirm concentration and purity; store in dark, desiccated, -20°C. |
| PDMS (Sylgard 184) | Dow, #4019862) | Elastomeric stamps or microfluidic devices for contact patterning or fluid delivery. | Strictly enforce 10:1 base:curing agent ratio; degas under consistent vacuum pressure/time. |
Protocol 2.1: Standardized Coating of PEG-passivated Substrates
- Glass Activation:
- Clean 25 mm #1.5 coverslips in piranha solution (3:1 H₂SO₄:H₂O₂) CAUTION: Extremely corrosive for 20 minutes.
- Rinse 5x in Milli-Q water, 2x in absolute ethanol, and dry under N₂ stream.
- Plasma treat at high setting for 5 minutes.
- PLL-PEG Adsorption:
- Immediately immerse coverslips in 0.1 mg/mL PLL-PEG solution in 10 mM HEPES (pH 7.4).
- Incubate for 45 minutes at room temperature in a sealed, dark container.
- Rinse 3x with sterile PBS, gently dry edges with lint-free wipe, and store in PBS at 4°C for up to 72 hours.
Protein-Ink Formulation & Handling
Protocol 2.2: Preparation of Light-Activatable Protein "Ink":
- Working Solution Prep (Day of Use):
- Thaw aliquots of protein of interest (e.g., fibronectin), 10x PBS, and LAP stock (50 mM in water) on ice.
- Prepare "Master Mix" in low-protein-binding tube: 90 µL of protein at 1.5x desired final concentration in 1x PBS, 10 µL of LAP stock. Final [LAP] = 5 mM.
- Mix by gentle inversion, centrifuge briefly, and keep on ice in light-blocking tube.
- Validation Spot Test:
- Apply 2 µL of the ink to a test acrylate-coated slide.
- Expose to 405 nm light (10 mW/cm², 5 seconds) through a simple photomask.
- Rinse vigorously with PBS+0.1% Tween-20, then image for patterned fluorescence. This confirms ink reactivity before primary experiment.
Instrument Calibration & Data Acquisition Standards
Table 3: Critical Calibration Schedule for LIMAP Equipment
| Instrument | Calibration Metric | Procedure & Frequency | Acceptance Criterion |
|---|---|---|---|
| 405 nm LED/Laser System | Irradiance (mW/cm²) | Measure with calibrated photodiode sensor at sample plane. Before each experiment. | Variation < ±5% from setpoint. |
| Micropipettes (µL range) | Volume Accuracy/Gravimetric | Dispense 10 aliquots of distilled water; weigh. Monthly. | CV < 1.0% for volumes ≥10 µL; < 2.5% for volumes <10 µL. |
| Confocal Microscope | Axial Calibration (Z-step) | Image sub-resolution fluorescent beads through Z-stack. Quarterly. | Measured step size within 1% of commanded value. |
| Plate Reader | Fluorescence Intensity | Read stable fluorophore (e.g., Fluorescein) in all wells. Weekly. | Inter-well CV < 3%. |
Protocol 3.1: Daily LED Patterning System Readiness Check
- Power & Uniformity:
- Place photodiode at the center of the sample stage. Record power reading.
- Move sensor to 4 corners of the typical patterning field. Record readings.
- Acceptance:
- The central reading must be within 5% of the value logged at last full calibration.
- Corner readings must be within 15% of the center reading (indicating field uniformity).
- Documentation:
- Log all values in the instrument logbook. Do not proceed if out of spec.
Data & Metadata Reporting Framework
For each LIMAP experiment, the following metadata must be archived alongside raw images and analysis code:
- Environmental Logs (Temp, Humidity for experiment duration).
- Reagent Lot Numbers (Protein, LAP, PEG, etc.).
- Instrument Calibration Certificates/Logs.
- Exact Software Versions (Fiji, MATLAB, etc.) and analysis parameter files.
Visualizations
Title: LIMAP Experimental Workflow
Title: Four Pillars of LIMAP Reproducibility
Title: LIMAP Photopatterning Mechanism Steps
Validating LIMAP Patterns and Comparing Techniques for Your Research Needs
Within the context of developing the novel LIMAP (Light-Induced Molecular Adsorption Protein) micropatterning protocol, rigorous quality control is paramount. LIMAP enables the precise spatial organization of proteins on a substrate using controlled light exposure to induce selective adsorption. This article details the integrated validation methods—fluorescence microscopy, atomic force microscopy (AFM), and immunostaining—essential for characterizing pattern fidelity, surface topography, and biomolecular functionality, respectively. These combined techniques ensure the reliability and reproducibility of LIMAP-generated microenvironments for downstream applications in cell mechanobiology and high-content drug screening.
Validation Methodologies: Application Notes & Protocols
Fluorescence Microscopy for Pattern Fidelity Analysis
Application Note: Fluorescence microscopy is the primary method for rapid, qualitative, and quantitative assessment of protein adsorption patterns generated by LIMAP. It confirms the spatial resolution, contrast, and uniformity of the patterned fluorescently tagged proteins (e.g., fibronectin-Alexa Fluor 555).
Protocol: Quantitative Analysis of Patterning Efficiency
- Sample Preparation: Perform LIMAP protocol using a fluorescently conjugated protein (e.g., 50 µg/mL fibronectin-Alexa Fluor 488 in PBS) on a glass-bottom dish. Include a non-illuminated control region.
- Imaging: Acquire images using an epifluorescence or confocal microscope with a 20x or 40x objective. Use identical exposure time, gain, and laser power across all samples.
- Quantitative Analysis:
- Open images in ImageJ/Fiji.
- Define rectangular regions of interest (ROIs) over patterned and non-patterned (background) areas.
- Measure mean fluorescence intensity (MFI) for each ROI.
- Calculate Patterning Contrast Ratio (PCR) as:
PCR = (MFI_pattern - MFI_background) / MFI_background. - Calculate Coefficient of Variation (CV) of MFI across multiple patterned features to assess uniformity.
Table 1: Typical Fluorescence Microscopy QC Metrics for LIMAP Patterns
| Metric | Formula/Description | Target Value for QC | Measurement Tool |
|---|---|---|---|
| Patterning Contrast Ratio (PCR) | (MFI_pattern - MFI_background) / MFI_background |
> 10 (High contrast) | ImageJ |
| Feature Uniformity (CV) | (Std. Dev. of MFI across features / Mean MFI) x 100% |
< 15% | ImageJ |
| Spatial Resolution (µm) | Measured via line profile across a pattern edge (10%-90% intensity) | As designed (e.g., 5 ± 0.5 µm) | ImageJ |
| Pattern Fidelity | Visual comparison to photomask design | No breaks or merging | Confocal Microscopy |
Atomic Force Microscopy (AFM) for Topographical and Mechanical Validation
Application Note: AFM provides nanoscale resolution of the topographical features created by protein adsorption in LIMAP. It verifies monolayer formation, measures pattern height, and can map local adhesion forces, ensuring the protocol does not induce undesirable protein aggregation or surface damage.
Protocol: Topography and Force Spectroscopy on LIMAP Patterns
- Sample Preparation: Use LIMAP to pattern an unlabeled protein (e.g., collagen I) on a flat, rigid substrate like mica or silicon wafer. Rinse and store in PBS.
- AFM Imaging: Use tapping mode in liquid (PBS) with a sharp silicon nitride tip (spring constant ~0.1 N/m). Scan patterned and unpatterned areas (e.g., 10 µm x 10 µm).
- Data Analysis:
- Flatten scan data using AFM software.
- Use line section analysis to measure the step height of the protein pattern.
- Perform force spectroscopy measurements on and off the pattern. Approach-retract cycles (n=100 per region) yield force-distance curves.
- Analyze curves to derive adhesion force and effective Young's modulus using appropriate models (e.g., Hertz model).
Table 2: AFM Quantitative Parameters for LIMAP QC
| Parameter | Description | Expected Outcome for Monolayer | Instrument |
|---|---|---|---|
| Pattern Height (nm) | Step height from substrate to pattern top. | 1-5 nm (monomeric layer) | AFM, Tapping Mode |
| Surface Roughness (Rq) | RMS roughness within a patterned zone. | < 1 nm (smooth monolayer) | AFM Software |
| Mean Adhesion Force (pN) | Average pull-off force on pattern vs. off. | Higher on protein pattern | AFM, Force Spectroscopy |
| Protein Layer Stiffness (kPa) | Apparent Young's modulus from fitting. | Characteristic of native protein | AFM, Force Mapping |
Immunostaining for Biofunctional Validation
Application Note: Immunostaining confirms the biological identity, conformation, and accessibility of the patterned proteins. It is critical for validating that the light-induced adsorption process in LIMAP does not denature proteins or mask key epitopes needed for subsequent cell receptor binding.
Protocol: Immunofluorescence Validation of Patterned Protein Function
- Blocking & Staining: After LIMAP patterning (with unlabeled protein), block the sample with 3% BSA for 1 hour. Incubate with a primary antibody specific for the patterned protein (e.g., anti-fibronectin, 1:500 in 1% BSA) for 2 hours at RT.
- Washing & Secondary Detection: Wash 3x with PBS. Incubate with a fluorescent secondary antibody (e.g., Alexa Fluor 647 anti-rabbit, 1:1000) for 1 hour at RT, protected from light.
- Imaging & Analysis: Image using appropriate fluorescence channels. Colocalization analysis (e.g., Pearson's coefficient) between the immunostain signal and a fiduciary marker (if using dual-tagged protein) confirms biofunctionality.
Table 3: Immunostaining QC Metrics for Biofunctional Validation
| Metric | Method of Assessment | Acceptance Criterion |
|---|---|---|
| Antibody Specificity | Stain unpatterned substrate and pattern. | Signal only on pattern. |
| Epitope Accessibility | Compare stain intensity to reference (spray-coated protein). | > 80% of reference MFI. |
| Colocalization Fidelity | Calculate Manders' or Pearson's coefficient vs. direct fluorescent tag. | Coefficient > 0.85. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for LIMAP Validation
| Item | Function in Validation | Example Product/Catalog Number |
|---|---|---|
| Fluorescently Conjugated Protein | Direct visualization of patterned features. | Fibronectin, Alexa Fluor 555 Conjugate (Thermo Fisher, FNFG555) |
| High-Affinity Primary Antibodies | Immunostaining for biofunctional check. | Anti-Fibronectin [IST-4] (Abcam, ab6328) |
| Cross-linked Secondary Antibodies | Detection in immunostaining with minimal background. | Goat anti-Mouse IgG (H+L) Cross-Adsorbed, Alexa Fluor 647 (Invitrogen, A-21236) |
| BSA (Fraction V) | Blocking agent to reduce non-specific binding. | Bovine Serum Albumin (Sigma, A7906) |
| AFM Cantilevers for Liquid | High-resolution topography and force measurements in PBS. | Bruker ScanAsyst-Fluid+ tips (k~0.7 N/m) |
| Glass-Bottom Culture Dishes | Optimal substrates for high-resolution microscopy. | MatTek Dish, No. 1.5 coverglass (P35G-1.5-14-C) |
| PBS (pH 7.4), Sterile | Standard buffer for washing and dilution. | Dulbecco's Phosphate Buffered Saline (Gibco, 14190144) |
Visualized Workflows & Pathways
Title: Integrated LIMAP Quality Control Validation Workflow
Title: Step-by-Step Immunostaining Protocol for Biofunctional QC
Title: Key Data Outputs and Metrics from AFM Validation
Within the broader thesis on advancing Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning protocols, this document establishes standardized Application Notes and Protocols for quantifying the three pillars of pattern fidelity: feature size accuracy, edge sharpness, and protein density. These metrics are critical for researchers, scientists, and drug development professionals creating high-fidelity protein patterns for applications in cell mechanobiology, multiplexed immunoassays, and organ-on-a-chip systems.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent/Material | Function in LIMAP Fidelity Quantification |
|---|---|
| Poly(ethylene glycol)-silane (PEG-silane) | Forms the non-fouling, protein-resistant background. Quality directly impacts edge sharpness. |
| Photo-reactive biotin conjugate (e.g., Benzophenone-PEG-Biotin) | The key "ink" for LIMAP. Adsorbs upon UV exposure, enabling streptavidin-protein tethering. |
| Streptavidin, Alexa Fluor 647 Conjugate | Standardized fluorescent probe for quantifying adsorbed protein density via fluorescence intensity. |
| Fluorescently-labeled target protein (e.g., Fibronectin, Alexa Fluor 488) | Model protein for assessing functional patterning fidelity and density. |
| Phosphate Buffered Saline (PBS) with 0.05% Tween 20 | Standard washing buffer to remove non-specifically adsorbed proteins without damaging patterns. |
| Poly-D-Lysine coated or plasma-treated glass substrates | Provides a uniform, positively charged surface for robust PEG-silane monolayer formation. |
| Calibrated UV Light Source (e.g., 365 nm LED) | Must have a homogenizer and intensity meter. Exposure dose controls feature size and density. |
| Atomic Force Microscopy (AFM) tips (TESPA-V2 type) | For high-resolution topography scans to measure feature height and edge roughness. |
Table 1: Typical Fidelity Metrics for Optimized LIMAP Protocol
| Metric | Measurement Method | Target Value (Optimal) | Common Range | Key Influencing Factor |
|---|---|---|---|---|
| Minimum Reliable Feature Size | Fluorescence microscopy line profile | 2 µm | 2 - 5 µm | UV mask resolution, optical blur, surface diffusion |
| Edge Sharpness (10-90% Intensity Rise) | Fluorescence microscopy line profile | 0.8 µm | 0.7 - 1.5 µm | UV coherence, photo-bleaching, wash stringency |
| Patterned Protein Density | Fluorescence intensity calibration vs. known standards | 4000 molecules/µm² | 3000 - 6000 molecules/µm² | UV dose, biotin linker concentration, incubation time |
| Background Protein Adsorption | Fluorescence intensity in non-irradiated areas | < 5% of pattern intensity | 2 - 10% | PEG-silane monolayer quality, blocking step efficacy |
| Pattern Thickness/Height | Atomic Force Microscopy (AFM) | 6 nm | 4 - 8 nm | Protein monolayer packing, streptavidin-biotin layers |
Table 2: Impact of UV Exposure Dose on Fidelity Parameters
| UV Dose (J/cm² @ 365 nm) | Effective Feature Size vs. Mask (%) | Edge Sharpness (10-90% rise, µm) | Relative Protein Density (%) |
|---|---|---|---|
| 0.5 | +15% (Over-spread) | 1.5 | 65% |
| 1.0 (Recommended) | +3% (Nominal) | 0.8 | 100% (Reference) |
| 2.0 | -8% (Under-spread) | 1.2 | 120% |
| 4.0 | -25% (Severe shrinkage) | 2.0 | 95% (Potential degradation) |
Experimental Protocols
Protocol 1: Quantifying Feature Size and Edge Sharpness via Fluorescence Microscopy
Objective: To measure the deviation of patterned features from the photomask design and to quantify the abruptness of pattern boundaries. Materials: LIMAP-patterned sample with fluorescently-labeled streptavidin, epifluorescence or confocal microscope with calibrated stage, image analysis software (e.g., ImageJ/FIJI). Procedure:
- Image the patterned sample using a 40x or 60x objective lens, ensuring the camera is not saturated.
- For line patterns, draw a perpendicular line profile across at least 5 different feature edges.
- For spot patterns, perform a radial average intensity analysis.
- Feature Size: Report the Full Width at Half Maximum (FWHM) for each profile. Compare the average to the mask dimension.
- Edge Sharpness: For each profile, identify the positions where intensity rises from 10% to 90% of its maximum. The distance between these points is the 10-90% rise distance. Report the mean and standard deviation across all measurements. Note: Use a low-concentration fluorescent dye in mounting media to confirm no optical aberrations at the measurement locations.
Protocol 2: Quantifying Patterned Protein Density via Fluorescence Calibration
Objective: To determine the absolute number of protein molecules adsorbed per unit area within the pattern. Materials: Alexa Fluor 647-labeled streptavidin of known labeling ratio, LIMAP substrate patterned with biotin, fluorescence microscope with stable light source, reference slides with known fluorophore density (or a calibration curve generated using serial dilutions of free dye in polymer film). Procedure:
- Incubate the patterned substrate with a saturating concentration (e.g., 10 µg/mL) of the labeled streptavidin for 15 minutes. Wash thoroughly.
- Acquire an image using consistent exposure time, gain, and laser power.
- Convert fluorescence intensity values (in arbitrary units) from the image to molecular density using the calibration curve. The calibration must account for the microscope's detection efficiency and the fluorophore's quantum yield.
- Correct for non-specific binding by subtracting the average background fluorescence from a non-irradiated, PEG-coated area treated identically.
- Report density in molecules/µm², specifying the fluorescent probe used.
Protocol 3: Assessing Topographical Fidelity via Atomic Force Microscopy (AFM)
Objective: To measure the three-dimensional height and surface roughness of protein patterns, complementing fluorescence data. Materials: LIMAP-patterned sample (can be unlabeled), AFM with tapping mode capability, sharp silicon nitride tips (k ~40 N/m). Procedure:
- Mount the sample securely on the AFM stage. Operate in tapping mode in liquid (PBS) to prevent dehydration artifacts.
- Scan areas encompassing both patterned and non-patterned regions. Recommended scan size: 20 µm x 20 µm to 5 µm x 5 µm.
- Analyze height profiles. Measure the step height between the patterned feature and the PEG background.
- Analyze line profiles to assess edge sharpness topographically. The root-mean-square (RMS) roughness of the patterned region should be calculated and compared to the background.
- Report mean feature height (nm) and RMS roughness (nm) for both patterned and background areas.
Supporting Diagrams
Diagram 1: LIMAP Workflow & Fidelity Metrics
Diagram 2: LIMAP Chemistry & Fidelity Impact
Within the broader research on the LIMAP (Light-Induced Molecular Adsorption Protein Micropatterning) protocol, functional validation of the fabricated protein patterns is paramount. LIMAP enables high-resolution, user-defined immobilization of extracellular matrix (ECM) proteins and bioactive ligands onto cell culture substrates using selective light activation. This application note details standardized protocols for assessing the biological efficacy of these patterns, specifically evaluating cell adhesion, spreading, and subsequent bioactivity. These validation steps are critical for applications in fundamental cell biology, engineered tissue models, and targeted drug development screens.
Research Reagent Solutions Toolkit
| Item | Function in Validation |
|---|---|
| Fibronectin, Laminin, Collagen I | ECM proteins commonly patterned via LIMAP to provide specific integrin-binding sites and study adhesion dynamics. |
| Fluorescently-Tagged Phalloidin | Binds filamentous actin (F-actin), enabling visualization and quantification of cell spreading and cytoskeletal organization. |
| Hoechst 33342 or DAPI | Cell-permeable nuclear stains for identifying individual cells and quantifying cell number/adhesion density. |
| Phospho-Specific Antibodies (e.g., p-FAK, p-ERK) | Immunostaining reagents to assess early signaling pathway activation downstream of patterned ligand engagement. |
| Calcein-AM / Ethidium Homodimer-1 | Live/Dead viability assay components for assessing cytotoxicity of the patterning process or subsequent treatments. |
| Blocking Solution (e.g., 1% BSA or serum) | Used to passivate non-patterned areas, confining cell attachment exclusively to the protein patterns. |
| Trypsin-EDTA (0.05%) | Standardized enzyme solution for controlled cell detachment in adhesion strength assays. |
| PDMS Stamps or Gaskets | Used to create defined wells on patterned substrates for localized seeding and assay application. |
Core Validation Protocols
Protocol: Quantitative Cell Adhesion Assay on Patterns
Objective: To quantify the number of cells selectively adherent to LIMAP-generated protein patterns versus non-patterned areas over time. Materials: LIMAP-patterned substrate, cell suspension, complete medium, PBS, 4% paraformaldehyde (PFA), Hoechst 33342, fluorescence microscope with automated stage. Procedure:
- Blocking: Incubate the patterned substrate with 1% BSA in PBS for 1 hour at 37°C to block protein adsorption on non-irradiated areas.
- Cell Seeding: Seed cells at a low density (e.g., 5,000 cells/cm²) in serum-free or low-serum medium to minimize non-specific adhesion. Allow cells to adhere for a predetermined time (e.g., 30, 60, 120 min).
- Washing: Gently rinse the substrate 3x with pre-warmed PBS to remove non-adherent cells.
- Fixation & Staining: Fix cells with 4% PFA for 15 min, permeabilize with 0.1% Triton X-100 (optional), and stain nuclei with Hoechst 33342 (1 µg/mL) for 10 min.
- Imaging & Analysis: Automatically acquire images from ≥10 pattern replicates and adjacent non-patterned areas. Use image analysis software (e.g., ImageJ, CellProfiler) to count nuclei within pattern boundaries and in control areas. Data Output: Adhesion efficiency calculated as (Cells on Pattern / Total Cells Seeded) or as a ratio (Pattern/Background).
Protocol: Cell Spreading and Morphometric Analysis
Objective: To measure the degree of cell spreading and cytoskeletal organization confined to the pattern geometry. Materials: LIMAP-patterned substrate, adherent cells, PBS, PFA, Triton X-100, fluorescent phalloidin, Hoechst, confocal or high-resolution fluorescence microscope. Procedure:
- Cell Culture on Patterns: Seed and culture cells on blocked patterns for 4-24 hours to allow full spreading.
- Fixation & Permeabilization: Fix with 4% PFA for 15 min. Permeabilize with 0.1% Triton X-100 in PBS for 5 min.
- Cytoskeletal Staining: Incubate with fluorescent phalloidin (1:500) and Hoechst in PBS for 30-60 minutes protected from light.
- Imaging: Acquire high-resolution z-stack images of the actin cytoskeleton and nucleus.
- Morphometric Analysis: Using thresholding and segmentation, quantify for each cell:
- Area: Total cell footprint.
- Aspect Ratio: Major axis / Minor axis.
- Circularity: 4π(Area/Perimeter²).
- Actin Alignment: Orientation relative to pattern axis. Data Output: Mean and distribution of morphometric parameters per pattern shape/size.
Protocol: Assessment of Focal Adhesion and Early Signaling Activation
Objective: To validate the bioactivity of patterned ligands by detecting phosphorylation events in key adhesion and growth signaling pathways. Materials: LIMAP-patterned substrate, cells, PBS, PFA, quenching solution (e.g., 100mM Glycine), blocking/permeabilization buffer (5% normal serum, 0.3% Triton), primary antibodies (anti-pFAK[Y397], anti-pERK1/2), fluorescent secondary antibodies, mounting medium. Procedure:
- Stimulation: Serum-starve cells for 4-6 hours post-adhesion to patterns. Stimulate with a growth factor or keep in starvation medium as a control for 5-15 min.
- Rapid Fixation: Fix cells immediately with 4% PFA for 15 min to preserve phosphorylation states. Quench with 100mM Glycine.
- Immunostaining: Block and permeabilize for 1 hour. Incubate with primary antibody overnight at 4°C. Wash and incubate with secondary antibody for 1 hour at RT.
- Imaging & Quantification: Acquire images using consistent settings. Measure mean fluorescence intensity (MFI) of phospho-signal within the cell footprint or at the cell-substrate interface (focal adhesions). Normalize to total protein or cell number.
Data Presentation
Table 1: Representative Quantitative Outcomes from LIMAP Pattern Validation
| Validation Metric | Pattern Type (Example) | Measurement Method | Typical Result (Example) | Key Implication |
|---|---|---|---|---|
| Adhesion Specificity | 20µm Fibronectin Lines | Nuclei Count (Pattern vs. Background) | Ratio > 50:1 | Excellent confinement by LIMAP and blocking. |
| Spreading Morphology | 30µm x 30µm Fibronectin Squares | Cell Area / Circularity | Area: ~900µm² ± 50; Circularity: 0.85 ± 0.05 | Cells conform precisely to pattern boundaries. |
| Early Signaling | 15µm Laminin Dots | p-FAK MFI per Cell | 2.5-fold increase over BSA background | Patterned ligand actively engages integrin signaling. |
| Proliferation | Grid of Collagen I | EdU+ Cells after 24h | Pattern-dependent alignment of division axis | Patterns can direct cell fate spatially. |
| Viaibility | Any LIMAP Pattern | Live/Dead Stain (Calcein/EthD-1) | >95% Viability | LIMAP process is non-cytotoxic. |
Visualization of Workflow and Signaling
Workflow for Functional Validation of LIMAP Patterns
Key Signaling Pathways Activated by Patterned Ligands
This analysis is presented within the framework of a doctoral thesis investigating the Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning protocol. The thesis aims to develop LIMAP as a versatile, user-friendly alternative to established techniques like microcontact printing (µCP) for generating high-fidelity protein patterns to control cell behavior. This document provides a comparative application note for researchers in cell biology, mechanobiology, and drug development.
Table 1: Qualitative Comparison of LIMAP and µCP
| Feature | LIMAP (Light-Induced Molecular Adsorption) | Microcontact Printing (µCP) |
|---|---|---|
| Principle | Photochemical activation of an inert surface (e.g., PLL-g-PEG) via projected UV light to selectively adsorb proteins. | Physical stamping of proteins from an elastomeric PDMS stamp onto a substrate. |
| Resolution | High (~1-10 µm), limited by optics and photochemistry. | Very High (sub-500 nm), limited by stamp deformation and master fabrication. |
| Pattern Flexibility | Extremely High. Digital pattern change without new hardware. | Low. Requires fabrication of a new physical stamp for each pattern. |
| Multipatterning | Excellent. Sequential, aligned patterning of different proteins is straightforward. | Challenging. Requires precise alignment of multiple stamps, risk of contamination. |
| Protein Compatibility | Broad; any protein in solution can be adsorbed. Risk of UV denaturation if directly exposed. | Broad; gentle transfer. Stamped proteins can dry or denature on stamp. |
| Throughput | Moderate for single patterns; high for multi-protein complex patterns. | High for replicating a single pattern many times. |
| Substrate Topography | Limited to flat, optically accessible surfaces for projection. | Can be adapted for mildly curved or non-planar surfaces. |
| Equipment Need | Requires UV light source, digital micromirror device (DMD) or mask, and optics. | Requires master fabrication (photolithography), PDMS, and plasma cleaner. |
| Key Advantage | Digital, maskless, rapid prototyping of complex, multi-protein patterns. | High resolution, well-established, low equipment cost for replication. |
| Key Limitation | Specialized optical setup, potential for non-specific adsorption in non-irradiated areas. | Stamp collapse for high-aspect-ratio features, pattern inflexibility after stamp fabrication. |
Table 2: Quantitative Performance Metrics
| Parameter | LIMAP | µCP | Notes |
|---|---|---|---|
| Typical Feature Size | 5 - 50 µm | 0.5 - 100 µm | µCP excels at submicron features. |
| Patterning Time | 1-10 min (exposure + incubation) | 10-60 min (stamp incubation + stamping) | Excludes setup/fabrication time. |
| Protein Consumption | Low (µL volumes in droplet) | Moderate (stamp incubation volume) | |
| Pattern Alignment Accuracy | ~ ±1 µm (digital) | ~ ±5-10 µm (manual) | Depends on equipment stage precision. |
| Cell Adhesion Efficiency | 85-95% (on patterns) | 80-95% (on patterns) | Varies with protein and passivation. |
Detailed Experimental Protocols
Protocol A: LIMAP Patterning for Focal Adhesion Studies
Objective: Create alternating 20 µm lines of fibronectin and vitronectin on a single substrate to study competitive cell adhesion.
Materials: See "The Scientist's Toolkit" below.
Procedure:
- Substrate Preparation: Clean a glass-bottom dish with ethanol and plasma treat for 2 minutes.
- Passivation: Incubate with 0.1 mg/mL PLL-g-PEG in HEPES buffer for 30 min. Rinse thoroughly with PBS.
- First Protein Patterning:
- Place a droplet of fibronectin solution (50 µg/mL in PBS) on the passivated surface.
- Project the first UV light pattern (parallel 20 µm lines) for 60-120 seconds.
- Aspirate the protein solution and rinse 3x with PBS.
- Second Protein Patterning:
- Without moving the sample, place a droplet of vitronectin solution (50 µg/mL in PBS).
- Project the second, complementary UV pattern (offset lines) for 60-120 seconds.
- Aspirate and rinse 3x with PBS.
- Cell Seeding: Trypsinize cells, resuspend in serum-free medium, and seed at low density onto the patterned substrate. Allow to adhere for 1-2 hours before adding complete medium.
Protocol B: µCP Patterning of Collagen Islands
Objective: Stamp isolated 50 µm x 50 µm squares of collagen I onto a polystyrene dish for single-cell confinement studies.
Materials: PDMS (Sylgard 184), SU-8 silicon master, collagen I (rat tail, 0.1 mg/mL in acetic acid), oxygen plasma cleaner.
Procedure:
- Stamp Fabrication: Pour PDMS (10:1 base:curing agent) over the SU-8 master. Cure at 65°C for 2 hours. Peel off, cut stamps, and use a biopsy punch to create a relief channel.
- Stamp Inking: Incubate the stamp face-down in a droplet of collagen I solution for 1 hour in a humid chamber.
- Stamp Drying: Gently dry the inked stamp under a stream of nitrogen.
- Substrate Activation: Treat a polystyrene culture dish with oxygen plasma for 30 seconds.
- Stamping: Briefly bring the dry, inked stamp into conformal contact with the activated substrate. Apply gentle, even pressure for 30 seconds. Peel the stamp away carefully.
- Passivation: Immediately incubate the stamped dish with 1% Pluronic F-127 in PBS for 1 hour to block non-patterned areas.
- Cell Seeding: Rinse with PBS and seed cells as in Protocol A.
Visualization of Workflows and Signaling Context
Diagram 1: LIMAP Experimental Workflow (5 Steps)
Diagram 2: Microcontact Printing Workflow (7 Steps)
Diagram 3: Thesis Focus: From LIMAP Patterns to Cell Response
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for LIMAP Protocol Development
| Item | Function in Experiment | Example/Notes |
|---|---|---|
| PLL(20)-g[3.5]-PEG(2) | Forms the inert, non-fouling monolayer on positively charged substrates. UV-cleavable PEG branches enable local photopatterning. | Commercial source: SuSoS AG; Critical parameter: grafting ratio. |
| DMD-based UV Projector | Generates the digital, high-resolution UV light pattern (~365 nm) for selective surface activation. | Can be custom-built or commercial. Key parameter: irradiance (mW/cm²). |
| Glass-bottom Culture Dishes | Provide an optically clear, flat, and modifiable substrate for patterning and live-cell imaging. | Often coated with SiO₂ or TiO₂ for reliable PLL-g-PEG adsorption. |
| Extracellular Matrix Proteins | The bioactive ligands patterned to direct cell behavior (e.g., adhesion, signaling). | Fibronectin, Collagen I, Laminin, Vitronectin. Must be in PBS (no amines). |
| Pluronic F-127 or BSA | Used as a blocking agent after patterning to ensure complete passivation of non-patterned areas. | Post-pattern incubation step to minimize non-specific adhesion. |
| HEPES Buffer (pH 7.4) | Optimal buffer for the PLL-g-PEG coating step, providing ionic strength without interfering amines. | Preferable over PBS for the passivation step. |
Within the broader thesis on Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning protocol research, this application note provides a comparative analysis of LIMAP against established inkjet and extrusion bioprinting technologies. The focus is on critical operational parameters—resolution, speed, and scalability—that define their applicability in biofabrication and drug development research.
Quantitative Comparison of Core Metrics
Table 1: Comparative Analysis of Micropatterning and Bioprinting Technologies
| Parameter | LIMAP | Inkjet Bioprinting | Extrusion Bioprinting |
|---|---|---|---|
| Spatial Resolution | 1 - 10 µm | 10 - 50 µm | 100 - 1000 µm |
| Printing Speed | ~10 mm²/s (for patterning) | 1 - 10,000 droplets/s | 1 - 50 mm/s (extrusion speed) |
| Scalability (Build Area) | Moderate (chip-scale) | High (platform-scale) | Very High (large constructs) |
| Cell Viability | >95% (post-patterning) | 85 - 95% | 60 - 95% (pressure-dependent) |
| Typical Bioink Viscosity | N/A (protein solutions) | 3.5 - 12 mPa·s | 30 - 6x10⁷ mPa·s |
| Multimaterial Capability | Sequential, layer-by-layer adsorption | Good (multiple printheads) | Good (multiple extruders) |
| Key Advantage | Ultra-high resolution, molecular control | High speed, good cell viability | Structural integrity, broad material use |
Detailed Experimental Protocols
Protocol 1: Standard LIMAP Micropatterning Workflow
Note: This protocol is central to the thesis research on light-induced molecular adsorption.
Materials:
- Substrate: Gold-coated glass slide (with Self-Assembled Monolayer, SAM)
- Protein Solution: 50 µg/mL fibronectin in PBS
- Photoactivatable Linker: Carbene-generating diazirine compound (e.g., SDSD-1065)
- Light Source: 365 nm UV LED array (5 mW/cm²)
- Phosphate Buffered Saline (PBS), pH 7.4
Procedure:
- Substrate Preparation: Clean gold-coated slides in piranha solution (3:1 H₂SO₄:H₂O₂) for 10 minutes. Rinse with deionized water and ethanol. Incubate in 1 mM alkanethiol solution (e.g., EG6) for 24 hours to form a SAM.
- Linker Coating: Incubate the SAM-functionalized slide in a 1 mM solution of the diazirine crosslinker in ethanol for 1 hour. Rinse thoroughly with ethanol and dry under nitrogen.
- Mounting and Alignment: Mount the slide on the LIMAP stage. Using the alignment software, define the target micropattern (e.g., 5 µm lanes, 10 µm spacing).
- Light Patterning: Pipette 100 µL of the fibronectin solution onto the slide surface. Activate the UV LED array with the defined pattern for 30-60 seconds. The light induces carbene formation, covalently binding fibronectin to the illuminated areas.
- Quenching and Washing: Incubate the slide in a 1% (w/v) bovine serum albumin (BSA) solution for 1 hour to block non-specific adsorption. Wash three times with PBS.
- Validation: Visualize patterned protein using fluorescence-tagged antibodies or direct tagging (e.g., FITC). Image with a confocal microscope.
Protocol 2: High-Throughput Inkjet Bioprinting of Cell-Laden Droplets
Materials: Thermal or piezoelectric inkjet printer, low-viscosity bioink (alginate/collagen with cells), crosslinking solution (CaCl₂).
Procedure:
- Bioink Preparation: Suspend cells (e.g., NIH/3T3) in 3% (w/v) alginate solution at a density of 1-5x10⁶ cells/mL. Keep on ice.
- Printer Calibration: Load bioink into cartridge. Perform nozzle priming and test firing to ensure consistent droplet formation (typically 1-100 pL/droplet).
- Printing: Execute the print G-code to deposit droplets in the desired 2D pattern (e.g., a grid) onto a petri dish.
- Crosslinking: Immediately after printing, mist the structure with 100 mM CaCl₂ solution to ionically crosslink alginate.
- Culture: Transfer to incubator, add culture medium. Assess cell viability via live/dead assay at 24 hours.
Protocol 3: Extrusion Bioprinting of a 3D Perfusable Channel
Materials: Pneumatic or screw-driven extruder bioprinter, gelatin methacryloyl (GelMA) bioink, photoinitiator (LAP), 405 nm light source.
Procedure:
- Bioink Synthesis: Synthesize 10% (w/v) GelMA. Dissolve 0.25% (w/v) Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator.
- Printer Setup: Load bioink into a temperature-controlled syringe (maintained at 15-20°C). Equip a 22G conical nozzle.
- Printing Parameters: Set pressure to 25-35 kPa, printing speed to 10 mm/s. Print a tubular structure (2 layers high, 8 mm diameter) directly into a support bath or onto a cooled stage.
- Photocrosslinking: After printing, expose the entire structure to 405 nm light (10 mW/cm²) for 60 seconds.
- Post-processing: If using a support bath, gently remove and transfer the structure to culture medium.
Visualization of Workflows and Relationships
Diagram 1: LIMAP experimental workflow
Diagram 2: Technology selection logic
Diagram 3: LIMAP molecular mechanism
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Featured Experiments
| Item | Function/Application | Example Product/Catalog |
|---|---|---|
| Diazirine Crosslinkers | Photoactivatable linker for LIMAP; forms reactive carbenes upon UV exposure. | Sigma-Aldrich, SDSD-1065 |
| Alkanethiols (e.g., EG6) | Forms self-assembled monolayers (SAMs) on gold substrates for LIMAP. | ProChimia, TH 001-06 |
| Recombinant Fibronectin | High-purity extracellular matrix protein for creating cell-adhesive micropatterns. | Gibco, 33016015 |
| Low-Viscosity Alginate | Biopolymer for inkjet bioinks; enables gentle cell encapsulation and rapid crosslinking. | NovaMatrix, SLG100 |
| Gelatin Methacryloyl (GelMA) | Photocrosslinkable hydrogel for extrusion bioprinting; provides tunable mechanical properties. | Advanced BioMatrix, GEL-IC-50 |
| Lithium Phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) | Highly efficient water-soluble photoinitiator for visible light crosslinking (405 nm). | TCI Chemicals, L0276 |
| Fluorescent Conjugates (FITC, TRITC) | For tagging proteins or antibodies to visualize and validate micropatterns post-fabrication. | Thermo Fisher Scientific |
| Piezoelectric Printhead | Core component of non-thermal inkjet bioprinters for precise droplet generation. | MicroFab, MJ-ABL-01 |
| Temperature-Controlled Syringe | Maintains bioink viscosity during extrusion printing for consistent deposition. | Nordson EFD, 7018252 |
This application note is framed within a broader thesis research program focused on advancing Light-Induced Molecular Adsorption Protein (LIMAP) micropatterning. The core thesis investigates LIMAP's mechanism, optimizes its protocol for robustness and accessibility, and critically evaluates its performance against established techniques like microfluidic photopatterning via immobilization (µPIV). The objective is to provide a definitive resource for selecting and implementing protein patterning methods in cell biology and drug development.
Table 1: Core Characteristics of Photopatterning Methods
| Feature | LIMAP | µPIV | Deep UV Patterning |
|---|---|---|---|
| Principle | Localized, light-induced adsorption of proteins onto azobenzene-containing polymer films. | UV-driven cleavage of photocleavable linker (e.g., NBPC) within a microfluidic channel to immobilize protein. | UV ozone oxidation of non-fouling coatings (e.g., PEG-silane) to create adhesive regions. |
| Resolution | ~1-5 µm | ~5-50 µm (channel width dependent) | ~1-10 µm |
| Throughput | High (parallel patterning via projection) | Low (serial patterning per channel) | Moderate (mask-based, batch) |
| Multiplexing | Excellent (in situ, sequential patterning without channels) | Limited (requires re-configuration of channel network) | Poor (typically single protein) |
| Substrate Flexibility | High (any azobenzene-coated substrate) | Restricted (requires bonded PDMS microfluidic device) | High (glass, silicon) |
| Equipment Complexity | Moderate (requires UV light source & mask/projector) | High (requires microfabrication & microfluidic control) | Low (requires UV ozone cleaner & mask) |
| Key Advantage | Dynamic, multi-component patterns without physical barriers. | Precise temporal control and fluid exchange during patterning. | Simple, widely accessible protocol. |
| Key Limitation | Requires synthesis of specialized photoresponsive polymer. | Low throughput, multiplexing is cumbersome. | Lack of in situ multiplexing, potential for non-specific adsorption. |
Table 2: Quantitative Performance Metrics
| Metric | LIMAP | µPIV | Source / Context |
|---|---|---|---|
| Patterning Time (for 5 patterns) | ~5-10 min (parallel) | ~25-50 min (serial) | Based on standard protocol durations. |
| Feature Alignment Accuracy | ± 0.5 µm | ± 2-5 µm (channel deformation) | Instrumental and soft lithography limits. |
| Protein Activity Retention | >90% (via ELISA) | >95% (gentle cleavage) | Assayed post-immobilization. |
| Pattern Stability (in cell media) | >72 hours | >96 hours (protected by channel) | Measured via fluorescence intensity. |
| Minimum Feature Size Demonstrated | 1 µm | 5 µm | Published experimental results. |
Detailed Experimental Protocols
Protocol 1: LIMAP for Multi-Component Patterning This protocol is central to the thesis research on LIMAP optimization.
Materials:
- Substrate: Glass coverslip coated with azobenzene-containing polymer (e.g., PAH-azo).
- Proteins: Fluorescently labeled proteins (e.g., Fibronectin-Alexa 488, Laminin-Alexa 594) in PBS.
- Equipment: UV light source (365 nm, 10 mW/cm²), photomask or digital micromirror device (DMD), humid chamber.
Procedure:
- Substrate Preparation: Place the PAH-azo coated substrate in a humid chamber.
- First Protein Adsorption: Apply a solution of the first protein (e.g., 50 µg/mL Fibronectin-488 in PBS) as a droplet on the substrate. Incubate for 1 min in dark.
- First Patterning: Align Photomask #1. Expose to UV light for 60-120 seconds. Light exposure causes trans to cis isomerization, inducing local protein adsorption.
- Rinse: Gently rinse the substrate with PBS to remove unbound protein. Blot edges dry.
- Second Protein Adsorption: Apply the second protein solution (e.g., Laminin-594).
- Second Patterning: Align Photomask #2. Expose to UV light. The cis-isomer regions from step 3 are now non-adhesive, directing adsorption to new areas.
- Final Rinse & Storage: Rinse thoroughly with PBS and store in PBS at 4°C until use (within 24 hrs).
Protocol 2: µPIV for Spatially-Controlled Immobilization
Materials:
- Microfluidic Device: PDMS device bonded to glass, pre-filled with heterobifunctional photoreactive crosslinker (e.g., NBPC-azide).
- Protein: Target protein modified with DBCO/cyclooctyne group.
- Equipment: UV laser (355 nm) or filtered mercury lamp, microfluidic pressure controller.
Procedure:
- Channel Preparation: Flow NBPC solution through channels, incubate, then rinse to create a uniform photocleavable monolayer.
- Protein Loading: Introduce the DBCO-modified protein solution. Allow diffusion but do not immobilize yet.
- Photopatterning: Using the UV laser, illuminate specific regions of the channel according to the desired pattern. This cleaves the NBPC linker, revealing an azide group that undergoes rapid strain-promoted click reaction with the DBCO on the protein.
- Channel Rinse: Flush the channel extensively with buffer to remove all unclicked, non-specifically adsorbed protein.
- Device Disassembly (Optional): The PDMS slab can be peeled away, leaving the patterned protein on the glass substrate for downstream cell culture.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Photopatterning
| Item | Function | Example Product/Chemical |
|---|---|---|
| PAH-azo Polymer | LIMAP Substrate Coating. Contains azobenzene chromophores for light-induced switching. | Synthesized in-house via diazo-coupling of PAH and Disperse Red 1. |
| NBPC Crosslinker | µPIV Photocleavable Linker. Forms the immobilized, photocleavable monolayer for protein capture. | (2-Nitrobenzyl) 3-(4-azidophenyl)propyl carbonate. |
| DBCO-modified Protein | µPIV Protein Handle. Enables specific, bioorthogonal click chemistry immobilization post-cleavage. | Protein-DBCO conjugate (commercial kits available). |
| High-Resolution Photomask | Defines the UV exposure pattern for LIMAP/Deep UV. | Chrome-on-quartz mask (≈ 2µm features). |
| Digital Micromirror Device (DMD) | For dynamic, maskless LIMAP patterning. | DLP LightCrafter 6500. |
| PDMS & Curing Agent | Fabrication of microfluidic channels for µPIV. | Sylgard 184 Silicone Elastomer Kit. |
| UV Light Source (365 nm) | Activating light for both LIMAP (trans->cis) and µPIV (cleavage). | UV LED System (e.g., Thorlabs). |
Pathway and Workflow Visualizations
Diagram 1: LIMAP Sequential Patterning Workflow (80 chars)
Diagram 2: LIMAP vs µPIV Core Mechanism (65 chars)
Diagram 3: Method Selection Logic Tree (55 chars)
The development of novel biomaterial interfaces is critical for advancing fundamental cell biology and therapeutic discovery. Within this context, the LIMAP (Light-Induced Molecular Adsorption Protein) micropatterning protocol research represents a significant advancement, offering high-resolution, dynamic, and cytocompatible spatial control over protein presentation. This framework positions LIMAP within the broader landscape of surface patterning techniques, providing researchers with a structured decision-making tool to select the optimal method for their specific biological question or application in drug development.
Patterning Technique Comparison and Quantitative Data
The choice of patterning technique is governed by key parameters: resolution, throughput, material compatibility, cost, and the ability to create dynamic patterns. The following table summarizes the primary techniques.
Table 1: Comparative Analysis of Key Protein Patterning Techniques
| Technique | Mechanism | Best Resolution | Dynamic Pattern Changes? | Typical Cost (Setup) | Key Advantages | Major Limitations | Ideal For |
|---|---|---|---|---|---|---|---|
| Microcontact Printing (µCP) | PDMS stamp transfer | ~1 µm | No | Low | Simple, fast, high-throughput, cell-compatible inks. | Static patterns only; limited to simple geometries; master fabrication required. | High-throughput static adhesion studies; co-culture initiation. |
| Inkjet Printing | Non-contact droplet deposition | 50-100 µm | Yes (by design) | Medium | Digital control, multi-material deposition, moderate scalability. | Lower resolution; potential nozzle clogging; splashing. | Medium-resolution multi-factor arrays; gradient printing. |
| Photolithography | UV light through a photomask | <1 µm | No (without complex steps) | Very High | Ultra-high resolution, industrial scalability. | Requires cleanroom; harsh chemicals (photoresists, developers); not directly cell-friendly. | Microelectronic integration; ultra-high density features. |
| Electron Beam Lithography | Focused e-beam writing | <10 nm | No | Extremely High | Unparalleled resolution. | Very slow, extremely expensive, requires vacuum. | Nanoscale protein features for fundamental biophysics. |
| LIMAP (Light-Induced Molecular Adsorption) | Photoactivation of adhesive surfaces (e.g., PEG) | ~5 µm | Yes (In Situ) | Medium | Live-cell compatible, spatiotemporal control, good resolution, uses standard microscopy. | Requires photoactivatable surfaces; potential phototoxicity at high doses. | Studying cell signaling dynamics; migration; reversible adhesion; synthetic tissue morphogenesis. |
Detailed Experimental Protocols
Protocol A: Standard Microcontact Printing (µCP) for Fibronectin Patterns
Objective: To create static islands of fibronectin for single-cell adhesion studies.
- Master Fabrication: Generate a photomask with desired features (e.g., 20 µm circles). Use photolithography to create a silicon wafer master.
- PDMS Stamp Preparation: Mix Sylgard 184 elastomer base and curing agent (10:1 ratio). Pour over master, degas, and cure at 65°C for 2+ hours. Peel off and cut stamps.
- Ink Application: Incubate stamp with 50 µg/mL fibronectin in PBS for 1 hour. Rinse with water and dry with nitrogen stream.
- Stamp Incubation: Place inked stamp in contact with a tissue culture dish for 1-2 minutes.
- Backfilling: Incubate stamped dish with 1% Pluronic F-127 in PBS for 30+ minutes to passivate non-patterned areas. Rinse with PBS before cell seeding.
Protocol B: Core LIMAP Workflow for Dynamic Patterning
Objective: To spatially control the adsorption of a protein of interest (e.g., RGD peptide) using light in the presence of cells.
- Substrate Preparation: Coat a glass-bottom dish with a photoactivatable copolymer (e.g., PLPP). Incubate for 1 hour, then rinse thoroughly to create a non-fouling background.
- Cell Seeding (Optional): Seed cells in a serum-free or low-serum medium. They will remain largely non-adherent on the passivated surface.
- Pattern Definition & Photoactivation: Using a digital micromirror device (DMD) or confocal microscope, project a defined pattern of 365-405 nm light onto the substrate. Typical dose: 0.5-2 J/cm².
- Protein Adsorption: Immediately introduce the solution containing the protein of interest (e.g., 10 µg/mL RGD-Cy3 in PBS) to the medium. Adsorption occurs selectively on illuminated regions within minutes.
- Live-Cell Imaging & Dynamic Changes: Image cell adhesion and response. To alter the pattern, illuminate new areas and introduce new proteins, enabling study of dynamic cellular reactions.
Visualizing the Decision Framework and LIMAP Mechanism
Decision Tree for Patterning Technique Selection
LIMAP Mechanism: Light-Induced Protein Adsorption
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for LIMAP and Related Patterning Experiments
| Item | Function/Benefit | Example/Note |
|---|---|---|
| Photoactivatable Coating | Forms the non-adhesive background that becomes locally adhesive upon UV exposure. | PLPP Copolymer: Poly(L-lysine)-graft-poly(ethylene glycol) with photocleavable o-nitrobenzyl groups. |
| Digital Micromirror Device (DMD) | Enables fast, flexible, and high-resolution pattern projection without physical masks. | Integrated into systems like Mosaic (Andor) or built into modern microscopes. |
| Bioinactive Passivant | Blocks non-specific protein adsorption on non-illuminated areas. | Pluronic F-127: Used in µCP. For LIMAP, the PLPP itself is the passivant. |
| ECM Protein/Peptide | The biological signal presented to cells. | Fibronectin, Collagen I, RGD-containing peptides. Can be fluorescently tagged for visualization. |
| PDMS (Sylgard 184) | Elastomer for creating stamps in microcontact printing. | Gold standard for µCP due to its elasticity and gas permeability. |
| High-Resolution Photomask | Defines the pattern in photolithography and µCP master fabrication. | Chrome-on-quartz for high fidelity; can be printed for lower resolution. |
| Glass-bottom Culture Dishes | Provide optimal optical clarity for high-resolution live-cell imaging during/after patterning. | Essential for microscopy-based techniques like LIMAP. |
Conclusion
LIMAP light-induced molecular adsorption protein micropatterning is a robust, versatile, and highly precise technique that empowers researchers to engineer sophisticated cellular microenvironments. By mastering its foundational principles, following a detailed optimized protocol, and applying systematic troubleshooting, scientists can reliably create complex protein patterns to direct cell behavior with unprecedented control. The validation and comparative analysis underscore LIMAP's unique position, offering a compelling blend of non-contact operation, high resolution, and compatibility with diverse biomolecules. As we look to the future, LIMAP is poised to play a pivotal role in advancing fundamental cell biology, developing more physiologically relevant organ-on-a-chip and tissue engineering models, and enabling next-generation high-content drug screening platforms. Its adaptability promises to keep pace with the growing demand for sophisticated tools to decipher and manipulate cell-microenvironment interactions in biomedical research.